
Abstract
Glioblastoma multiforme (GBM) is the most malignant and common brain tumor; it is aggressive growth pattern means that GBM patients face a poor prognosis even when receiving the best available treatment modalities. In recent years, an increasing number of reports suggest that the discovery of microRNAs (miRNAs) might provide a novel therapeutic target for human cancers, including GBM. One miRNA in particular, microRNA-25 (miR-25), is overexpressed in several cancers, wherein accumulating evidence indicates that it functions as an oncogene. However, the function of miR-25 in GBM has not been totally elucidated. In this study, we demonstrated that miR-25 was significantly up-regulated in astrocytoma tissues and glioblastoma cell lines. In vitro studies further demonstrated that overexpressed miR-25 was able to promote, while its antisense oligos inhibited cell proliferation and invasion in U251 cells. Moreover, we identified neurofilament light polypeptide (NEFL) as a novel target molecule of miR-25. Also of note was the fact that NEFL was down-regulated with increased levels of miR-25 expression in human astrocytoma clinical specimens. In addition, via the mTOR signaling pathway, NEFL-siRNA could significantly attenuate the inhibitory effects of knockdown miR-25 on the proliferation and invasion of U251 cells. Overall, our results showed an important role for miR-25 in regulating NEFL expression in GBM, and suggest that miR-25 could be a potential target for GBM treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Glioblastoma multiforme (GBM), the most malignant form of glioma, is highly aggressive and neurologically destructive [1]. Thanks to the increasing body of knowledge about this malignancy at genetic and molecular levels, and the considerable advances in cancer therapy, patient outcomes have slowly improved over the past decade [1]. Despite this progress, however, the prognosis and survival rates for patients are still extremely poor [2], and the average life expectancy of patients diagnosed with GBM is still only 14 months [3]. Hence, there is an urgent need to explore novel agents for the treatment of this highly aggressive disease, as well as to better understand the molecular and cellular alterations that occur in GBM.
MicroRNAs (miRNAs) are a group of small, non-coding 18- to 25- nucleotide-long RNAs that play important roles in cancer development by regulating the activities of specific mRNA targets [4]. They are aberrantly expressed in many cancers and can exert tumor suppressive or oncogenic functions [5]. Aberrant expression of miRNAs in glioblastoma has also been reported, and many miRNAs have been shown to participate in glioblastoma tumorigenesis and/or invasion by targeting oncogenes or tumor suppressor genes, including miR-96, miR-101, MiR-125a-5p, miR-128, miR-182, miR-185, and miR-381 [6–15]. These findings indicate that miRNAs may act as another kind of important regulator in glioblastoma tumorigenesis, in addition to the protein-coding genes.
miR-25 was reported as an oncogenic miRNA involved in many tumor types, such as gastric cancer, prostate cancer, lung cancer, ovarian cancer, colorectal cancer, hepatocellular carcinoma, breast cancer, etc. [16–25]. However, the role of miR-25 in glioblastoma tumorigenesis and the underlying molecular mechanisms by which miR-25 exerts its functions had remained—until recently—largely unknown. Here, we demonstrated that miR-25 is critical in regulating glioblastoma cell proliferation and invasion and also boosts glioma tumorigenesis. Moreover, miR-25 was aberrantly significantly up-regulated in astrocytoma tissues and cell lines. A mechanistic study revealed that miR-25 regulated glioblastoma tumorigenesis by modifying the targets of NEFL via the mTOR signaling pathway.
Results
miR-25 was overexpressed in human astrocytoma samples and cell lines
To analyze the expression levels of miR-25 in human astrocytoma samples, we first profiled miRNA expression in 44 astrocytoma samples and 20 normal brain tissues using the quantitative RT-PCR technique. Our analysis showed that the expression levels of miR-25 were significantly higher in the astrocytoma samples compared with the normal brain tissues (Fig. 1a). Furthermore, we examined the expression of miR-25 in glioblastoma cell lines. We tested a panel of three brain cell lines, including one cell line derived from normal human astrocytes (NHA) as control. The results showed that miR-25 was overexpressed in the glioblastoma cell lines U251, LN229, and U87 (Fig. 1b). This evidence indicated that miR-25 may be involved in glioblastoma tumorigenesis.

miR-25 was overexpressed in human glioma tumor samples and cell lines. a qRT-PCR analysis showing that expression of miR-25 is increased in astrocytomas as compared with that in the normal brain tissues. b qRT-PCR analysis showing that expression of miR-25 is increased in glioblastoma cell lines as compared with that in the normal human astrocytes (NHA). The data represent the mean ± SDs of 3 replicates. ***p < 0.001
The expression of miR-25 could affect the proliferation and invasion of glioblastoma cells
In order to explore the biological significance of miR-25 in glioblastoma tumorigenesis, we up- (Fig. 2a) or down-regulated (Fig. 2b) the expression of miR-25 in U251 cells. We then investigated the effect of miR-25 on the proliferation of U251 cells using a CCK-8 assay (Fig. 2c, d). As shown in Fig. 2c, when compared to control group cells, miR-25 overexpression by transfected with miR-25 mimics significantly increased the proliferation of U251 cells at 24 h post-transfection. Similarly, when miR-25 in U251 cells was knocked down using a miR-25 inhibitor, cell proliferation was inhibited at 24 h post-transfection when compared with control group cells (Fig. 2d). Next, a wound-healing assay, clonogenic assay, and a matrigel chamber invasion assay were performed. The wound-healing assay was used to detect the impact of miR-25 on cell motility. Overexpressed miR-25 promoted the migration and motility of U251 cells (Fig. 2e). Additionally, targeted inhibition of miR-25 with the miRNA inhibitor reduced the migration and motility of U251 cells(Fig. 2f). The clonogenic assay and matrigel chamber invasion assay also showed similar results. As shown in Fig. 2g, i, compared with that of the negative control, overexpressed miR-25 promotes cell colony formation and invasion in U251 cells (p < 0.001). Furthermore, knockdown of miR-25 could constrain cell colony formation (Fig. 2h) and invasion (Fig. 2j). These data provide strong evidence that knockdown of miR-25 could inhibit the proliferation, migration, and invasion in U251 cells, while overexpression of miR-25 could increase cell proliferation, migration, and invasion.

The expression of miR-25 could affect the proliferation and invasion of glioblastoma cells. a qRT-PCR analysis showing that expression of miR-25 is significantly increased in U251 cells upon miR-25 mimic transfection. b qRT-PCR analysis showing that expression of miR-25 is significantly decreased in U251 cells upon miR-25 inhibitor transfection. c CCK8 assay showing the increased proliferation of U251 cells transfected with miR-25 mimics. d CCK8 assay showing the reduced proliferation of U251 cells transfected with miR-25 inhibitor. e Overexpression of miR-25 promotes tumor cell migration, as determined by in vitro wound-healing assays. f Knockdown miR-25 inhibits tumor cell migration, as determined by in vitro wound-healing assays. g Colony formation assay showing the promoted cellular colony formation after U251 cells were transfected with miR-25 mimics. h Colony formation assay showing the reduced cellular colony formation after U251 cells were transfected with miR-25 inhibitor. i Matrigel chamber invasion assay showing promoted invasion of U251 cells after being transfected with miR-25 mimics. j Matrigel chamber invasion assay showing reduced invasion of U251 cells after being transfected with miR-25 inhibitor. The data represent the mean ± SDs of 3 replicates. *p < 0.05, **p < 0.01,***p < 0.001
NEFL is a new target molecule of miR-25
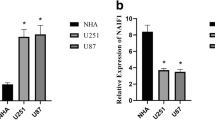
As shown in Fig. 3a, the miRNA target prediction program TargetScan6.2 was used to identify miR-25 interaction sites in the 3′-UTR of NEFL. Therefore, HEK293 and U251 cells were co-transfected with the wild-type (WT) or mutation-type (MT) NEFL luciferase reporter vector, together with miR-25 or the negative control miR-NC, for 24 h. Luciferase activity was significantly reduced in cells transfected with WT NEFL and miR-25, but not in the cells transfected with MT NEFL and miR-25 (Fig. 3b). We further determined the expression of NEFL by using Quantitative real-time (qRT) PCR (Fig. 3c) and western blotting analysis (Fig. 3d) in U251 cells. As shown in Fig. 3c and d, the expression of the NEFL mRNA and protein was down-regulated in miR-25-treated U251 cells but was increased in miR-25 knockdown cells. The previous study showed that NEFL binds to TSC1 and stabilize the TSC1/2 complex [26]. Down-regulation of NEFL has been shown to lead to abnormal activation of the mTOR pathway [27]. Moreover, NEFL sensitizes glioblastoma cells to TMZ by inhibiting the mTOR pathway [13]. Therefore, we hypothesized that the abnormal expression of miR-25 leads to mTOR pathway change via targeting of NEFL.

NEFL is a direct target of miR-25. a Schematic of the interaction sites of miR-25 in the 3′-UTRs of NEFL. b Luciferase assays of HEK293 and U251 cells co-transfected with pMIR-REPORT- WT/MT 3′-UTR NEFL and miR-25 or the negative control, as indicated. c qRT-PCR analysis showing the mRNA level of NEFL after miR-25 mimics or inhibitor was transfected into U251 cells for 24 h. miR-25 down-regulated the mRNA level of NEFL. d Western blot analysis showing the protein expression of NEFL, p-p70S6 K, and p70S6 K after the miR-25 mimics or inhibitor was transfected into U251 cells for 48 h. miR-25 decreased the protein expression of NEFL and p-p70S6 K; GAPDH was used as a loading control. e qRT-PCR analysis showing that the mRNA level of NEFL was significantly reduced in astrocytoma tissues compared to normal brain tissues. f Spearman’s correlation analysis was used to determine the correlation between the expression levels of NEFL and miR-25 in human astrocytomas; Spearman’s correlation, r = −0.639 (n = 44). g Spearman’s correlation analysis was used to determine the correlation between the expression levels of NEFL and miR-25 in human normal brain tissue; Spearman’s correlation, r = 0.150 (n = 20). The data represent the mean ± SDs of 3 replicates. ***p < 0.001
To test this hypothesis, we examined the status of the mTOR pathway and the effects of altered NEFL expression in U251 cell lines by overexpression of miR-25 (Fig. 3d). Consistent with our hypothesis, mTOR pathway activity was inhibited upon NEFL up-regulation. The phosphorylation of p70S6 k, a critical downstream substrate of activated mTOR and known indicator of an active mTOR pathway, was then analyzed in U251 cells (Fig. 3d). Restoration of NEFL expression by knockdown of miR-25 suppressed p70S6 k phosphorylation in U251 cells (Fig. 3d right), whereas down-regulation of NEFL expression by miR-25 resulted in increased phosphorylation of p70S6 k in U251 cells (Fig. 3d left). Then, we measured the expression levels of NEFL in normal brain tissues and astrocytoma samples. RT-qPCR assays showed that NEFL expression was significantly decreased in the astrocytoma samples (n = 44) when compared with the normal brain tissues (n = 20) (Fig. 3e). Because NEFL is a target molecule of miR-25, we also determined the correlation between NEFL and miR-25 in astrocytoma samples. We found an inverse correlation between the expression of miR-25 and the level of NEFL mRNA in the astrocytoma samples (Spearman’s correlation, r = −0.639) (Fig. 3f), but not in normal tissues (Spearman’s correlation, r = 0.150) (Fig. 3g). These results indicate that NEFL is a novel target of miR-25, and the abnormal expression of miR-25 leads to mTOR pathway change via targeting of NEFL.
NEFL-siRNA reverses the inhibitory effects of knockdown miR-25 in U251 cells via activating mTOR pathway
To further evaluate the contribution of NEFL to the biological effects of miR-25, we assessed the impact of NEFL knockdown by RNA interference. As shown in Fig. 4a, knockdown miR-25 with antisense oligos increased the expression of NEFL, and decreased the expression of phosphorylated p70S6 K, but not the total p70S6 K. When transfected with NEFL-siRNA to inhibit the expression of NEFL, the expression level of phosphorylated p70S6 K was reduced. These results indicated that miR-25 promoted the activation of the mTOR pathway by targeting NEFL. Subsequent studies showed the effect of NEFL knockdown on anti-miR-25-dependent cell proliferation and invasion in U251 cells (Fig. 4b–d). These data indicated that down-regulation of NEFL expression could significantly attenuate the inhibitory effect of miR-25 inhibitor on cell proliferation and invasion, suggesting that the down-regulated miR-25 inhibits the proliferation and invasion of U251 cells through the NEFL-mediated mTOR pathway.

NEFL-siRNA reverses the inhibitory effects of knockdown miR-25 in U251 cells. a Western blot analysis showing the protein expression level change of NEFL, phosphorylated p70S6 K, and total p70S6 K cells with the miR-25 inhibitor or NEFL-siRNA transfected in U251. b CCK8 assay showing the proliferation of U251 cells transfected with miR-NC or miR-25 inhibitor was forced to repress the expression of NEFL. The results showed that transfected miR-25 inhibitor decreased the proliferation of U251 cells. NEFL-siRNA attenuates the inhibitory effects of miR-25 inhibitor to U251 cells. c Cloning formation assay showing the reduced cellular cloning formation after U251 cells were transfected with miR-NC or miR-25 inhibitor was forced to repress the expression of NEFL. The results showed that transfected miR-25 inhibitor decreased the cloning formation of U251 cells. NEFL-siRNA attenuates the inhibitory effects of miR-25 inhibitor to U251 cells. d Matrigel of chamber invasion assay showing reduced invasion U251 cells after being transfected with miR-NC or miR-25 inhibitor was forced to repress the expression of NEFL. The results showed that transfected miR-25 inhibitor decreased the invasion of U251 cells. NEFL-siRNA attenuates the inhibitory effects of miR-25 inhibitor to U251 cells. The data represent the mean ± SDs of 3 replicates. Asterisks indicate a significant difference compared to the miR-NC + siR-NC group, Number sign indicates a significant difference compared to the miR-25 inhibitor + siR-NEFL group. *p < 0.05, **p < 0.01, ***p < 0.001, #p < 0.05, ##p < 0.01, ###p < 0.001
Discussion
GBM is characterized by high genetic heterogeneity and infiltrative brain invasion patterns. Over the last decade, accumulated evidence has shown that miRNAs play an important role in relevant molecular and cellular mechanisms governing GBM tumorigenesis [28]. Due to their small size and influence in a broad range of biological processes, miRNAs are very attractive therapeutic targets for GBM.
miR-25 is one of the most overexpressed miRNAs in a number of profiling experiments designed for the detection of miRNAs dysregulated in human cancers [16–25, 29]. Li et al. reported that miR-25 promoted gastric cancer migration, invasion, and proliferation by directly targeting transducer of ERBB2 and 1 and correlated with poor survival [17]. In ovarian cancer, miR-25 promotes cell proliferation and motility by targeting LATS2 [18]. Up-regulation of miRNA-25 in colorectal cancer and hepatocellular carcinoma was associated with patient poor prognosis [20, 29]. The present profile data further confirmed an increased expression of miR-25 both in astrocytoma samples and in cell lines. Furthermore, our in vitro studies revealed that overexpression of miR-25 promoted cell proliferation, migration, and invasion in U251 cells; similarly, knockdown miR-25 inhibited the proliferation, migration, and invasion. Therefore, our study indicates that miR-25 might be an oncogene in the progression of GBM.
NEFL is located on chromosome 8p21, which has been identified as a genetic locus frequently affected by both heterozygous and homozygous deletions in a variety of common human cancers, including prostate cancer [30], breast cancer [31, 32], and head and neck cancer (HNC) [27, 33]. NEFL, a potential tumor suppresser [30, 31, 33], is associated with resistance to chemosensitivity. Re-expression of NEFL in HNC significantly increases the sensitivity of the cells to the drug [27]. Overexpression of NEFL significantly suppressed the proliferation and invasion of U251 cells and enhanced the chemosensitivity of glioblastoma cells to TMZ [13].
Generally, miRNAs are believed to function in vivo by binding to the characteristic binding sites in the targets’ 3′-UTRs [4]. Furthermore, our data revealed that NEFL was a direct target of miR-25, and miR-25 down-regulated NEFL by binding to the 3′-UTR of NEFL mRNA; In addition, our results showed that miR-25 and NEFL were inversely correlated in human astrocytoma samples and normal brain tissues, which further confirmed that NEFL was a target gene of miR-25 and targeted by miR-25 for repression in glioblastoma cells. NEFL is an inhibitor of the mTOR pathway [26]. Up-regulation of NEFL by knockdown of miR-25 can increase expression of phosphorylated p70S6 K to activate the mTOR pathway. Down-regulation of NEFL could significantly attenuate the inhibitory effects of knockdown miR-25 on the proliferation, migration, and invasion of U251 cells via activation of the mTOR pathway. LATS2 and ERBB2 were also the direct targets of miR-25 [17, 18, 34]. LATS2 is a tumor suppressor gene of gastric adenocarcinoma (GAC) [34], malignant mesothelioma [35], ovarian cancer [18], breast cancer [36], non-small cell lung cancer [37], and prostate cancer [36]. Down-regulation of LATS2 promoted the growth and motility of cancer cells [35, 38]. In GAC cells and ovarian cancer, miR-25 regulated proliferation and invasion by targeting LATS2 [18, 34]. However, the role of LATS2 in glioblastoma tumorigenesis and the underlying molecular mechanisms had remained largely unknown. ERBB2, a known proto-oncogene, is located at the long arm of human chromosome 17 (17q12) [39], and overexpression of ERBB2 is a negative prognostic factor in anaplastic astrocytomas [40]. However, the relationship between miRNA and ERBB2 in GBM is remained largely unknown. So here we did not focus on the relationship between miR-25 and LATS2 or ERBB2.
In summary, we found that miR-25 was overexpressed in human astrocytoma samples and cell lines. Through knockdown and overexpression studies, we found that miR-25 promoted GBM cell proliferation and invasion. Moreover, miR-25 expression is inversely related with NEFL in human astrocytoma samples. miR-25 represses the activation of the mTOR pathway by targeting NEFL and inhibits GBM cell proliferation and invasion. Our data suggest that miR-25 could be a potential therapeutic agent for GBM.
Materials and methods
Human tissue samples
Human astrocytoma samples and normal brain tissues were obtained from the Department of Neurosurgery, Xiangya Hospital, Hunan, China. This study was approved by the hospital institutional review board, and written informed consent was obtained from all patients. All the protocols were reviewed by the Joint Ethics Committee of the Central South University Health Authority and performed following national guidelines. Tissue samples were collected at surgery, immediately frozen in liquid nitrogen, and stored until total RNAs or proteins were extracted.
Cell culture
NHA and Human glioblastoma cell lines (U87MG, U 251, and LN229) and Human Embryonic Kidney (HEK) 293 cells were maintained in DMEM medium with high glucose and sodium pyruvate, supplemented with 10 % fetal bovine serum and antibiotics (100 units/ml penicillin and 100 mg/ml streptomycin). Cells were incubated at 37 °C in a humidified atmosphere of 5 % CO2 in air.
RNA oligoribonucleotides and antibodies
The control RNA mimics, miR-25 mimics, control miRNA inhibitor, miR-25 inhibitor, and NEFL-siRNA were all obtained from GenePharma (Shanghai, China). The RNA oligoribonucleotide was transfected at a working concentration of 100 nM using Lipofectamine RNAiMAX reagent (Invitrogen) according to the manufacturer’s instructions. The miR-25 mimics sequence is 5′-CAUUGCACUUGUCUCGGUCU GA-3′. The control RNA mimics sequence is 5′-UCACAACCUCCUAGAAAGAGU AGA-3′. The miR-25 inhibitor sequence is 5′-CAUUGCACUUGUCUCGGUC UGA-3′. The control miRNA inhibitor sequence is 5′-UUUGUACUACACAAAAG UACUG-3′. The target sequence of the NEFL-siRNA is 5′-GUCCUACUACACCAG CCAUTT-3′. Antibodies against p70S6 K (#9202) and phospho-p70S6 K (#9204) were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibodies against NEFL (sc-20012) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA); antibodies against GAPDH (B661104-0001) were from sangon biotech(Sangon Biotech, shanghai, China). The detail information is as follows: (1) p70S6 K antibody: rabbit monoclonal antibody; molecular weight: 70KD; using range 1:2000 (WB). (2) phospho-p70S6 K antibody: rabbit monoclonal antibody; molecular weight: 70KD; using range 1:1000 (WB). (3) NEFL antibody: mouse monoclonal antibody; molecular weight: 66KD; using range 1:500 (WB). (4) GAPDH antibody: mouse monoclonal antibody; molecular weight: 36KD; using range 1:3000 (WB).
RNA extraction, reverse transcription, and real-time PCR analysis
Total RNAs were isolated from harvested cells or human tissues with Trizol reagent according to the manufacturer’s instruction (Invitrogen, CA, USA). RNAs were reverse transcribed using the PrimeScript™ RT-PCR Kit (Takara). Real-time PCR reactions were performed using SYBR Premix DimerEraser (Takara, Dalian, China). Human GAPDH or U6 snRNA was used as an endogenous control for mRNA or miRNA detection, respectively. Expression of each gene was quantified by measuring C t values and normalized using the 2-ΔΔct method relative to U6 snRNA or GAPDH. Primers used for miR-25 real-time PCR are forward, 5′-TCTGGTCTCCCTCACAG GAC-3′; reverse, 5′-CATGGGTCGCCTACTCAC-3′. Primers used for NEFL qRT- PCR are forward, 5′-CTGGAAATCGAAGCATGCCG-3′; reverse, 5′-CGCCTTCC AAGAGTTTCCTGT-3′.
Western blotting
Cells were washed with ice-cold PBS buffer, scraped from the dishes, and centrifuged at 1200 rpm, 4 °C for 15 min. Cell lysates were prepared using RIPA buffer supplemented with protease inhibitors (100 mM Tris (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1 % Triton X-100, 1 % deoxycholate acid, 0.1 % SDS, 2 mM phenylmethylsulfonyl fluoride, 1 mM sodium orthovanadate, 2 mM DTT, 2 mM leupeptin, and 2 mM pepstatin). The supernatants were collected, and protein concentration was determined using a BCA assay (Thermo, USA). Tumor tissues from humans were grinded into powder in liquid nitrogen with RIPA buffer, and the total tissue proteins were extracted as described above. Aliquots of protein lysates were fractionated by SDS-PAGE, transferred to a PVDF membrane (Merck Millipore,Germany), and subjected to immunoblotting analysis according to the manufacturer’s instruction. ECL Detection System (Merck Millipore, Germany) was used for signal detection.
Luciferase reporter assay
The 3′-UTR of NEFL was synthesized and annealed, then inserted into the MluI and HindIII sites of pMIR-reporter luciferase vector (Ambion) downstream of the stop codon of the gene for luciferase. For its mutagenesis, the sequences complementary to the binding site of miR-25 in the 3′-UTR (NEFL:GTGCAAT) were replaced by CACACAA. These constructs were validated by sequencing. The cells were seeded into a 24-well plate for luciferase assay. After being cultured overnight, cells were co-transfected with the wild-type or mutation-type plasmid, pRL-TK plasmid, and equal amounts of miR-25 or miR-NC; the pRL-TK control vector was transfected as a control. Luciferase assays were performed 24 h after transfection using the Dual Luciferase Reporter Assay System (Promega, WI, USA). Firefly and Renilla reniformis luciferase activities were measured 30 s later. Experiments were performed in three independent replicates.
Cell viability assay
Cell viability was determined by the CCK8 assay. Briefly, 2000 cells/well were seeded into 96-well plates and were treated by miRNA, and cell absorption was measured using a CCK8 kit (Beyotime Institute of Biotechnology, Jiangsu, China) according to the manufacturer’s instruction at different indicated time points. Data were from three separate experiments with four replications each time.
Matrigel chamber invasion assay
Invasion assay was determined using 24-well BD Matrigel invasion chambers (Corning Inc., Corning, NY) in accordance with the manufacturer’s instructions. 2 × 104 cells were seeded per well in the upper well of the invasion chamber in DMEM with 0.1 % serum, the lower chamber well contained DMEM supplemented with 10 % FBS to stimulate cell invasion. After incubation for 24 h, non-invading cells were removed from the top well with a cotton swab, while the bottom cells were fixed with 4 % paraformaldehyde, stained with 0.1 % crystal violet, and photographed in three independent fields for each well. Three independent experiments were conducted in triplicate.
Wound-healing assay
Cells were cultured until they reached 90 % confluence in 6-well plates. Cell layers were scratched using a 10 μL tip to form wounded gaps, washed with PBS twice, and cultured. The wounded gaps were photographed at different time points and analyzed by measuring the distance of migrating cells from five different areas for each wound.
Statistical analysis
All experiments were performed three times, and data were analyzed with GraphPad Prism 5 (La Jolla, CA, USA). The correlation between miR-25 expression and NEFL levels in tumor tissues was analyzed using Spearman’s rank test. Differences between the variables of the groups were tested using the Student’s t test or one-way ANOVA, using the SPSS 15.0 program. A p-value of < 0.05 was considered to indicate a statistically significant result.
References
Zhu Y, Parada LF (2002) The molecular and genetic basis of neurological tumours. Nat Rev Cancer 2:616–626. doi:10.1038/nrc866
Ricard D, Idbaih A, Ducray F, Lahutte M, Hoang-Xuan K, Delattre JY (2012) Primary brain tumours in adults. Lancet 379:1984–1996. doi:10.1016/S0140-6736(11)61346-9
Wen PY, Kesari S (2008) Malignant gliomas in adults. N Engl J Med 359:492–507. doi:10.1056/NEJMra07081264
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297. doi:10.1016/S0092-8674(04)00045-5
Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, Downing JR, Jacks T, Horvitz HR, Golub TR (2005) MicroRNA expression profiles classify human cancers. Nature 435:834–838. doi:10.1038/nature03702
Liu X, Lei Q, Yu Z, Xu G, Tang H, Wang W, Wang Z, Li G, Wu M (2015) MiR-101 reverses the hypomethylation of the LMO3 promoter in glioma cells. Oncotarget 6:7930–7943
She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, Luo Z, Li G, Wu M (2014) miR-181 subunits enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeleton remodeling in glioblastoma cells. Med Oncol 31:892. doi:10.1007/s12032-014-0892-9
She X, Yu Z, Cui Y, Lei Q, Wang Z, Xu G, Xiang J, Wu M, Li G (2014) miR-128 and miR-149 enhance the chemosensitivity of temozolomide by Rap1B-mediated cytoskeletal remodeling in glioblastoma. Oncol Rep 32:957–964. doi:10.3892/or.2014.3318
Tang H, Bian Y, Tu C, Wang Z, Yu Z, Liu Q, Xu G, Wu M, Li G (2013) The miR-183/96/182 cluster regulates oxidative apoptosis and sensitizes cells to chemotherapy in gliomas. Curr Cancer Drug Targets 13:221–231. doi:10.2174/1568009611313020010
Tang H, Liu X, Wang Z, She X, Zeng X, Deng M, Liao Q, Guo X, Wang R, Li X, Zeng F, Wu M, Li G (2011) Interaction of hsa-miR-381 and glioma suppressor LRRC4 is involved in glioma growth. Brain Res 1390:21–32. doi:10.1016/j.brainres.2011.03.034
Tang H, Wang Z, Liu Q, Liu X, Wu M, Li G (2014) Disturbing miR-182 and -381 inhibits BRD7 transcription and glioma growth by directly targeting LRRC4. PloS One 9:e84146. doi:10.1371/journal.pone.0084146
Tang H, Wang Z, Liu X, Liu Q, Xu G, Li G, Wu M (2012) LRRC4 inhibits glioma cell growth and invasion through a miR-185-dependent pathway. Curr Cancer Drug Targets 12:1032–1042. doi:10.2174/156800912803251180
Wang Z, Yang J, Xu G, Wang W, Liu C, Yang H, Yu Z, Lei Q, Xiao L, Xiong J, Zeng L, Xiang J, Ma J, Li G, Wu M (2015) Targeting miR-381-NEFL axis sensitizes glioblastoma cells to temozolomide by regulating stemness factors and multidrug resistance factors. Oncotarget 6:3147–3164
Yuan J, Xiao G, Peng G, Liu D, Wang Z, Liao Y, Liu Q, Wu M, Yuan X (2015) MiRNA-125a-5p inhibits glioblastoma cell proliferation and promotes cell differentiation by targeting TAZ. Biochem Biophys Res Commun 457:171–176. doi:10.1016/j.bbrc.2014.12.078
Zhang Z, Tang H, Wang Z, Zhang B, Liu W, Lu H, Xiao L, Liu X, Wang R, Li X, Wu M, Li G (2011) MiR-185 targets the DNA methyltransferases 1 and regulates global DNA methylation in human glioma. Mol Cancer 10:124. doi:10.1186/1476-4598-10-124
Li Z, Sun Y, Chen X, Squires J, Nowroozizadeh B, Liang C, Huang J (2015) p53 Mutation directs AURKA overexpression via miR-25 and FBXW7 in prostatic small cell neuroendocrine carcinoma. Mol Cancer Res 13:584–591. doi:10.1158/1541-7786.MCR-14-0277-T
Li BS, Zuo QF, Zhao YL, Xiao B, Zhuang Y, Mao XH, Wu C, Yang SM, Zeng H, Zou QM, Guo G (2014) MicroRNA-25 promotes gastric cancer migration, invasion and proliferation by directly targeting transducer of ERBB2, 1 and correlates with poor survival. Oncogene 34(20):2556–2565. doi:10.1038/onc.2014.214
Feng S, Pan W, Jin Y, Zheng J (2014) MiR-25 promotes ovarian cancer proliferation and motility by targeting LATS2. Tumour Biol 35:12339–12344. doi:10.1007/s13277-014-2546-0
Wang X, Wang HK, Li Y, Hafner M, Banerjee NS, Tang S, Briskin D, Meyers C, Chow LT, Xie X, Tuschl T, Zheng ZM (2014) microRNAs are biomarkers of oncogenic human papillomavirus infections. Proc Natl Acad Sci USA 111:4262–4267. doi:10.1073/pnas.1401430111
Li X, Yang C, Wang X, Zhang J, Zhang R, Liu R (2014) The expression of miR-25 is increased in colorectal cancer and is associated with patient prognosis. Med Oncol 31:781. doi:10.1007/s12032-013-0781-7
Kishikawa T, Otsuka M, Yoshikawa T, Ohno M, Takata A, Shibata C, Kondo Y, Akanuma M, Yoshida H, Koike K (2013) Regulation of the expression of the liver cancer susceptibility gene MICA by microRNAs. Sci Rep 3:2739. doi:10.1038/srep02739
Xu X, Chen Z, Zhao X, Wang J, Ding D, Wang Z, Tan F, Tan X, Zhou F, Sun J, Sun N, Gao Y, Shao K, Li N, Qiu B, He J (2012) MicroRNA-25 promotes cell migration and invasion in esophageal squamous cell carcinoma. Biochem Biophys Res Commun 421:640–645. doi:10.1016/j.bbrc.2012.03.048
Razumilava N, Bronk SF, Smoot RL, Fingas CD, Werneburg NW, Roberts LR, Mott JL (2012) miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death receptor-4 and promotes apoptosis resistance in cholangiocarcinoma. Hepatology 55:465–475. doi:10.1002/hep.24698
Poliseno L, Salmena L, Riccardi L, Fornari A, Song MS, Hobbs RM, Sportoletti P, Varmeh S, Egia A, Fedele G, Rameh L, Loda M, Pandolfi PP (2010) Identification of the miR-106b ~25 microRNA cluster as a proto-oncogenic PTEN-targeting intron that cooperates with its host gene MCM7 in transformation. Sci Signal 3:a29. doi:10.1126/scisignal.2000594
Yang T, Chen T, Li Y, Gao L, Zhang S, Wang T, Chen M (2015) Downregulation of miR-25 modulates non-small cell lung cancer cells by targeting CDC42. Tumour Biol 36:1903–1911. doi:10.1007/s13277-014-2793-0
Haddad LA, Smith N, Bowser M, Niida Y, Murthy V, Gonzalez-Agosti C, Ramesh V (2002) The TSC1 tumor suppressor hamartin interacts with neurofilament-L and possibly functions as a novel integrator of the neuronal cytoskeleton. J Biol Chem 277:44180–44186. doi:10.1074/jbc.M207211200
Chen B, Chen J, House MG, Cullen KJ, Nephew KP, Guo Z (2012) Role of neurofilament light polypeptide in head and neck cancer chemoresistance. Mol Cancer Res 10:305–315. doi:10.1158/1541-7786.MCR-11-0300
Costa PM, Cardoso AL, Nobrega C, Pereira DAL, Bruce JN, Canoll P, Pedroso DLM (2013) MicroRNA-21 silencing enhances the cytotoxic effect of the antiangiogenic drug sunitinib in glioblastoma. Hum Mol Genet 22:904–918. doi:10.1093/hmg/dds496
Su ZX, Zhao J, Rong ZH, Geng WM, Wu YG, Qin CK (2014) Upregulation of microRNA-25 associates with prognosis in hepatocellular carcinoma. Diagn Pathol 9:47. doi:10.1186/1746-1596-9-47
Macoska JA, Trybus TM, Benson PD, Sakr WA, Grignon DJ, Wojno KD, Pietruk T, Powell IJ (1995) Evidence for three tumor suppressor gene loci on chromosome 8p in human prostate cancer. Cancer Res 55:5390–5395
Knowles MA, Shaw ME, Proctor AJ (1993) Deletion mapping of chromosome 8 in cancers of the urinary bladder using restriction fragment length polymorphisms and microsatellite polymorphisms. Oncogene 8:1357–1364
Kang S, Kim B, Park SB, Jeong G, Kang HS, Liu R, Kim SJ (2013) Stage-specific methylome screen identifies that NEFL is downregulated by promoter hypermethylation in breast cancer. Int J Oncol 43:1659–1665. doi:10.3892/ijo.2013.2094
Coon SW, Savera AT, Zarbo RJ, Benninger MS, Chase GA, Rybicki BA, Van Dyke DL (2004) Prognostic implications of loss of heterozygosity at 8p21 and 9p21 in head and neck squamous cell carcinoma. Int J Cancer 111:206–212. doi:10.1002/ijc.20254
Zhang M, Wang X, Li W, Cui Y (2015) miR-107 and miR-25 simultaneously target LATS2 and regulate proliferation and invasion of gastric adenocarcinoma (GAC) cells. Biochem Biophys Res Commun 460:806–812. doi:10.1016/j.bbrc.03.110
Murakami H, Mizuno T, Taniguchi T, Fujii M, Ishiguro F, Fukui T, Akatsuka S, Horio Y, Hida T, Kondo Y, Toyokuni S, Osada H, Sekido Y (2011) LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res 71:873–883. doi:10.1158/0008-5472.CAN-10-2164
Li W, Wang L, Katoh H, Liu R, Zheng P, Liu Y (2011) Identification of a tumor suppressor relay between the FOXP3 and the Hippo pathways in breast and prostate cancers. Cancer Res 71:2162–2171. doi:10.1158/0008-5472.CAN-10-3268
Yao F, Liu H, Li Z, Zhong C, Fang W (2015) Down-regulation of LATS2 in non-small cell lung cancer promoted the growth and motility of cancer cells. Tumour Biol 36:2049–2057. doi:10.1007/s13277-014-2812-1
Yu T, Bachman J, Lai ZC (2015) Mutation analysis of large tumor suppressor genes LATS1 and LATS2 supports a tumor suppressor role in human cancer. Protein Cell 6:6–11. doi:10.1007/s13238-014-0122-4
Kodack DP, Askoxylakis V, Ferraro GB, Fukumura D, Jain RK (2015) Emerging strategies for treating brain metastases from breast cancer. Cancer Cell 27:163–175. doi:10.1016/j.ccell.2015.01.001
Gulati S, Ytterhus B, Granli US, Gulati M, Lydersen S, Torp SH (2010) Overexpression of c-erbB2 is a negative prognostic factor in anaplastic astrocytomas. Diagn Pathol 5:18. doi:10.1186/1746-1596-5-18
Acknowledgments
The authors thank Dr. Zeyou Wang for his precious suggestions and generous help in carrying out this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Peng, G., Yuan, X., Yuan, J. et al. miR-25 promotes glioblastoma cell proliferation and invasion by directly targeting NEFL. Mol Cell Biochem 409, 103–111 (2015). https://doi.org/10.1007/s11010-015-2516-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-015-2516-x