
Abstract
Hepatocyte cell adhesion molecule (HepaCAM) plays a crucial role in tumor progression and has been recognized as a novel tumor suppressor gene. The high protein expression level of protein kinase Cε (PKCε) has been discovered in many tumor types. In the present study, we determined HepaCAM and PKCε protein levels in human clear cell renal cell carcinoma (ccRCC) tissues and analyzed the correlation between them. We observed an inverse relationship in the expression of HepaCAM and PKCε in ccRCC and adjacent normal tissues. In ccRCC tissue, HepaCAM expression was undetectable while PKCε expression was high; the opposite was found in the adjacent normal tissue. Western blot analysis demonstrated that PKCε cytosolic protein levels increased while plasma membrane protein levels decreased without any change in total protein following infection of the ccRCC cell line 786-0 with adenovirus-GFP-HepaCAM (Ad-GFP-HepaCAM). Moreover, the application of Ad-GFP-HepaCAM combined with a PKCε-specific translocation inhibitor (εV1-2) effectively inhibited 786-0 cell growth. Ad-mediated expression of HepaCAM in 786-0 cells reduced the levels of phosphorylated AKT and cyclin D1 and inhibited cell proliferation. In summary, our studies point to interesting connections between HepaCAM and PKCε in tissues and in vitro. HepaCAM may prevent the translocation of PKCε from cytosolic to particulate fractions, resulting in the inhibition of 786-0 cell proliferation. Therapeutic manipulation of these novel protein targets may provide new ways of treating ccRCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Renal cell carcinoma accounts for 2–3 % of adult malignancies, and the incidence of this disease in developed countries is higher than in developing countries [1]. It has been known for years that clear cell renal cell carcinoma (ccRCC) is the most common type of kidney cancer, unfortunately most are not sensitive to traditional radiotherapy and chemotherapy. Approximately 20–40 % of patients undergoing nephrectomy develop multiple organ metastases [2]. Although therapeutic approaches have slowly improved over time, effective novel treatments for ccRCC are urgently needed.
Hepatocyte cell adhesion molecule (HepaCAM) was originally identified in hepatocellular cancer, and its mRNA level has been shown to be down-regulated in diverse cancer types [3–5]. Further investigation revealed that it functioned like a typical tumor suppressor gene, and that it was involved in many important signaling pathways [6, 7]. Interferon-γ has been shown to regulate HepaCAM expression levels in the human bladder cancer cell line, BIU-87, and promote G0/G1 phase arrest [8]. It has also been shown in cancer cell lines like MCF7, U373-MG, and T24 that exogenous expression of HepaCAM promotes cell cycle arrest [6, 9, 10]. Furthermore, it has been shown that exogenous expression of HepaCAM in 786-0 and RC-2 renal cancer cells was involved in c-Myc degradation [11].
The protein kinase C (PKC) family of serine/threonine kinases consists of at least 11 isoforms that contribute to cancer proliferation, apoptosis, metastasis, and drug-resistance [12–14]. The PKC family is made up of three major categories according to structure and biochemical features: the classical isoforms (α, βI, βII, and γ), the novel isoforms (δ, ε, η, θ, and μ), and the atypical isoforms (ζ and λ). Among them, PKCε, a transforming oncogene, has been recognized as a representative member of the novel isoforms that contains a C-terminal kinase catalytic element and an N-terminal regulatory region. There are two special regions at the N-terminus, C1 and C2. The C1 domain can influence protein–protein interactions and anchor PKCε to special subcellular membranes, which is determined by the cell type and second messengers, where it becomes an activated form [15, 16]. The C2 moiety of PKCε can mediate activated PKCε binding to its specific receptor called receptor for activated C kinase ε (RACKε), which is located in the cell membrane [17]. Furthermore, the PKCε translocation-specific inhibitor peptide, εV1-2, which is derived from the C2 fragment, is capable of targeting RACKε and hampering PKCε translocation [18].
Although much of the research has focused on key molecules involved in cancer cell growth that were regulated by means of HepaCAM, potential molecules that are associated with ccRCC growth have not been elucidated. Preliminary experiments conducted in our laboratory have suggested that exogenous expression of HepaCAM could upregulate BCL-2 and downregulate BAX in 786-0 cells (data not shown), which prompted us to take a closer look at PKCε. Its overexpression in many tumor types has been shown to modulate cell proliferation and apoptosis, and the activated form of PKCε was found to be associated with the cell membrane, similar to the location of HepaCAM as described previously [3, 19]. The purpose of our work was to clarify the molecular mechanisms responsible for the upregulation of HepaCAM, which resulted in the inhibition of proliferation of 786-0, a clear cell renal cancer cell line. Furthermore, we also investigated the effect of HepaCAM on PKCε intracellular redistribution in vitro, and whether HepaCAM could be a novel pharmacological target for treating this disease.
Materials and methods
Tissues
Fifteen human normal liver tissues were purchased from the department of tissue specimen database, and got all official licenses. Thirty-six tumor specimens and 36 adjacent non-cancerous tissue specimens were collected from patients seen in the Department of Urology, The First Affiliated Hospital of Chongqing Medical University (Chongqing, China) from 2012 to 2013. All patients underwent partial resection or radical nephrectomy, and samples were confirmed post-surgery using pathological examination according to the 2009 AJCC TNM-staging system and the 1997 WHO recommended Fuhrman four-grade system and pathological classification criteria of RCC [20, 21]. All specimens were stored in liquid nitrogen until use. Informed consent was obtained from all patients and the hospital’s Ethics Review Committee approved the study.
Cell line and culture conditions
The human ccRCC cell line, 786-0, was purchased from Ai Biological Research (Shanghai, China). 786-0 cells were cultured in RPMI 1640 medium (Life Technologies, Carlsbad, CA, USA) containing 10 % fetal calf serum (Life Technologies) at 37 °C in an incubator with a humidified atmosphere of 5 % CO2.
Immunohistochemistry
All tissues, upon removal from liquid nitrogen storage, were fixed in formalin and embedded in paraffin. Samples were cut on a microtome into 5 μm thick sections, deparaffinized in xylene, and rehydrated in graded alcohol. Endogenous peroxidase activity was blocked using a 3 % H2O2 solution. Sections were rehydrated in sodium citrate buffer and heated for 30 min for antigen retrieval. Sections were then incubated with normal goat serum diluted 1:10 in phosphate-buffered saline (PBS) for 1 h. Rabbit anti-HepaCAM polyclonal antibody (ProteinTech, Wuhan, China) and mouse anti-PKCε monoclonal antibody (BD Biosciences, San Jose, CA, USA) (dilution 1:200) were added to sections and incubated at 4 °C overnight. Sections were then treated with biotin-labeled anti-IgG and avidin-biotin horseradish peroxidase complex. Staining was carried out using diaminobenzidine (DAB), which was stopped when a brown color developed, and then counterstained with Mayer hematoxylin. The slides were then incubated in a reagent containing 99 % ethanol and 1 % hydrochloric acid and stained with lithium carbonate. Images were captured using microscopy and analyzed using IPP6.0 image analysis software. Percent positive staining and intensity were determined as described by Ruckhäberle et al. [22].
Immunoblot analysis
Adenovirus-GFP (Ad-GFP) and Adenovirus-GFP-HepaCAM (Ad-GFP-HepaCAM) were kind gifts from the Department of Laboratory Diagnosis, Chongqing Medical University. The specific PKCε translocation inhibitor, εV1-2 (AnaSpec Inc., Fremont, CA, USA), was used to study the function of PKCε translocation. 786-0 cells were subjected to εV1-2, Ad-GFP, and Ad-GFP-HepaCAM, individually or in combination after cells had attached to the bottom of the culture flask. Cells were harvested when green fluorescence could be seen in 70–80 % of cells in a random microscopic field, usually after 48 h. Cell total protein, plasma membrane, and cytosolic fractions were extracted using the Membrane and Cytosol Protein Extraction Kit (Beyotime Biotechnology, China) following the manufacturer’s protocol. Briefly, to extract total protein, cells were washed three times with PBS and then harvested in RIPA lysis buffer with added PMSF and phosphatase inhibitor (Na3VO4 and NaF) (Roche, Switzerland) on ice and vortexed every 10 min for 30 min. The lysed cells were centrifuged at 12,000 rpm for 30 min at 4 °C (Bio-Rad, USA), and the supernatant was removed. To extract membranes and cytosolic fractions, cells were lysed in isolation buffer (20 mM Tris–HCl, pH7.5 containing 10 mM MgCl2, 2 mM EGTA, 2 mM EDTA, 100 mM RIPA, 1 mM PMSF, 1 mM NaF, 1 mM Na3VO4, and 2 mM dithiothreitol) on ice for 15 min. To ensure that the cell samples were completely lysed, they were subjected to 3–5 freeze (liquid nitrogen)/thaw (room temperature) cycles until ~70 % of the cells were lysed as seen under light microscopy. The lysed cells were then centrifuged at 700×g for 10 min at 4 °C to eliminate nuclei and unlysed cells. The supernatant was collected (cytosolic fraction) after centrifugation at 14,000×g for 30 min at 4 °C. Finally, the sediment was lysed in buffer containing 1 % Triton X-100 for 15 min at 4 °C and centrifuged at 14,000×g for 5 min at 4 °C, which yielded the plasma membrane fraction in the supernatant. Protein concentration was measured using the BCA protein assay Kit (Beyotime Institute of Biotechnology, Shanghai, China). Equal amounts of membrane, cytosolic, and total extracts (120 μg) were prepared and subjected to SDS-polyacrylamide gel electrophoresis. Proteins were then transferred to a PVDF membrane and incubated with specific primary antibodies against HepaCAM, GAPDH (Proteintech, Wuhan, China), PKCε (BD Biosciences), Na+/K+-ATPase, phosphorylated AKT (p-AKT) and total AKT (Cell Signaling Technology, Beverly, MA, USA), cyclinD1, and β-actin (Santa Cruz Biotechnology, Dallas, TX, USA). Detection was carried out using HRP-conjugated secondary antibodies. Images were captured using an Odyssey scanner (LI-COR Biosciences, Lincoln, NE, USA) and analyzed using Quantity One software. Data were derived from three independent experiments.
CCK-8 assay
To explore the optimum concentration of εV1-2 and time point to use for inhibiting growth, cell proliferation was evaluated using the Cell Counting Kit-8 assay (7Sea Biotech, Shanghai, China). 786-0 cells were plated in 96-well plates at a density of 2,000 cells per well. εV1-2 was then added to the plates at concentrations of 0.5, 1, 5, 10 μM, and cultured for 3, 6, 12, 24, and 48 h after the cells had attached. A total of 10 μl of a CCK-8 solution was added to each well, and plates were incubated for an additional 3 h. Thereafter, O.D. was assessed by measuring the absorbance at 450 nm. Three independent experiments were run per condition.
Colony formation assay
Harvested cells were resuspended in RPMI 1640 medium and seeded in 6-well plates at ~500 cells per well, and then 24 h later, cells were treated with Ad-GFP, Ad-GFP-HepaCAM, and εV1-2 individually or in combination. Plates were incubated for 2 weeks in an incubator. Fresh culture medium was added one time after 10 days. After staining the cells with 0.005 % crystal violet, we counted the number of colonies containing more than 50 cells.
Statistical analyses
All statistical analyses were performed using SPSS 16.0 software, and all data are shown as the mean ± standard deviation (SD). HepaCAM and PKCε tissue protein levels were correlated using Pearson analysis. Comparison of data between two or more groups was carried out using t tests and one-way ANOVA. p value of less than 0.05 was considered significant.
Results
HepaCAM expression decreased while PKCε expression increased in ccRCC tissues
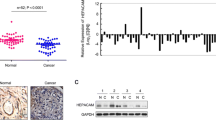
To investigate whether there was any correlation between HepaCAM and PKCε expression, we used anti-PKCε and anti-HepaCAM antibodies to detect the expression of PKCε and HepaCAM in 36 ccRCCs and adjacent tissues. Data showed that HepaCAM was weakly expressed in ccRCC tissues, but strongly expressed in adjacent tissues, especially in cytomembranes of kidney tubules (Fig. 1a, b; p < 0.05, Table 1). However, for PKCε, adjacent tissue expression levels were lower than in cancer tissues. Furthermore, we found that PKCε was more highly expressed in the cytoplasm and membrane, especially in cell-connected membranes, than in nuclei (Fig. 1c, d; p < 0.05, Table 1). In order to make it clear whether HepaCAM antibody is specific, we used human normal liver tissues served as control (Fig. 1e, f). We used the mean density for estimating the protein expression level of HepaCAM and PKCε. Results suggested that there was a negative linear correlation between HepaCAM and PKCε expression in the same patient according to Pearson analysis (r = −0.599, p < 0.05, Fig. 1i), and a higher expression level of PKCε was associated with a higher T stage and Fuhrman grade (p < 0.05, Table 1). However, there was no significant difference in age, sex, or disease recurrence.

The expression of HepaCAM and PKCε in clear cell renal cell cancer and adjacent nonmalignant tissues was determined using immunohistochemistry. HepaCAM was undetectable in clear cell cancer (a), but upregulated in adjacent normal samples, especially at the cytomembrane of kidney tubules (b). PKCε was found to be highly expressed in clear cell cancer tissues, especially in cell-connected membranes (c), but showed weak expression in adjacent tissues (d). Gray areas indicate positive. In human liver, HepaCAM expression served as positive control (e), liver PBS, renal cancer PBS, and HepaCAM note negative control (f–h). The original magnification was ×400. The correlation curve shows HepaCAM versus PKCε in ccRCC specimens using Pearson analysis (r = −0.599, p < 0.05, i)
Overexpression of HepaCAM-blocked PKCε translocation from cytoplasm to plasma membrane
To further explore the relationship between HepaCAM and PKCε in vitro, we infected 786-0 clear cell cancer cell lines with Ad-GFP-HepaCAM and investigated whether HepaCAM affected PKCε redistribution or total protein level. We extracted plasma membrane, cytoplasm, and total PKCε protein from cells expressing HepaCAM. Western blot analysis revealed that the 786-0 cell line was successfully infected with Ad-GFP-HepaCAM and Ad-GFP. The cytosolic protein level increased while the membrane protein level decreased remarkably in HepaCAM-overexpressing cells; however, total PKCε protein remained unchanged. Cell membrane β-actin blots uncovered the purity of membrane protein extractions, which should be expressed in cytoplasm substantially (p < 0.01, Fig. 2a, b). In short, HepaCAM could block PKCε redistribution from cytosolic to plasma membrane fractions in cells compared with blank and GFP controls.

HepaCAM exogenous expression could block PKCε translocation from the cytoplasm to the plasma membrane in 786-0 cells. Analysis of western blots indicated that HepaCAM was expressed successfully. After infection with Ad-GFP-HepaCAM, membrane protein was decreased, cytosolic protein was increased, but total PKCε protein was not affected by HepaCAM. Plasma membrane β-actin blots illustrate the high purity of membrane fractions. Results shown were from three independent experiments, the histogram of expression represent mean ± SD, with relative intensity normalized to β-actin and Na+/K+-ATPase individually, and with GFP and blank as controls (**p < 0.01, a, b)
HepaCAM and εV1-2 effectively inhibited ccRCC cell line proliferation
εV1-2 inhibited 786-0 cell proliferation in a concentration and time-dependent manner according to CCK-8 assay data. However, the highest inhibition rate was only about 40 % at 24 h (p < 0.05, Fig. 3a). We chose the optimum concentration of εV1-2 (10 μM) and Ad-GFP-HepaCAM with which to treat 786-0 cells for 14 days, and subsequently stained the cells with 0.005 % crystal violet. Results showed that after exposure to Ad-GFP-HepaCAM and εV1-2, cells had a significantly lower colony formation potential compared with cells infected only with Ad-GFP-HepaCAM (p < 0.05, Fig. 3b, c). However, εV1-2 could only partially inhibit clonal growth. We then examined p-AKT, total AKT, and cyclin D1, key proteins involved in cell cycle modulation, using western blotting. The data revealed that after treatment with Ad-GFP-HepaCAM and εV1-2, the levels of p-AKT and cyclin D1 were significantly lower compared with those in cells only infected with Ad-GFP-HepaCAM. Total AKT protein level, however, was not affected by the different conditions. We also found that εV1-2 alone could partially reduce the levels of p-AKT and cyclinD1, which may explain the observed effects on proliferation and colony formation (p < 0.05, Fig. 3d, e).

HepaCAM and εV1-2 were effective at inhibiting 786-0 cell growth. CCK-8 assay results revealed that the specific inhibitor of PKCε translocation, εV1-2, increased the 786-0 cell inhibition rate in a concentration and time-dependent manner. Time points included 3, 6, 12, 24, and 48. εV1-2 at 10 μM incubated for 24 h were the optimal concentration and time to use in the experiment (*p < 0.05, a). The number of colonies decreased substantially after exposure to Ad-GFP-HepaCAM and εV1-2 compared with Ad-GFP-HepaCAM alone, but εV1-2 alone had a partial effect on clone formation (b). Every colony with more than 50 cells was included in the analysis. The histogram illustrates the number of colonies per plate (*p < 0.05, c). Data from western blots show changes in p-AKT and cyclin D1 protein levels after cells were exposed to different conditions (d). The total AKT protein level was not changed. The bar graph indicates the relative intensity of each group, with quantified expression normalized to GAPDH (e) (*p < 0.05)
Discussion
In this study, we determined HepaCAM and PKCε expression profiles in ccRCC tissues and showed that HepaCAM could block PKCε translocation from cytosolic to cell plasma membrane fractions after infection of the 786-0 clear cell renal cancer cell line with Ad-GFP-HepaCAM. Interestingly, cells infected with Ad-GFP-HepaCAM and treated with εV1-2 at the same time were more strongly growth-inhibited than when cells were infected with Ad-GFP-HepaCAM alone.
As mentioned above, deletion of the HepaCAM gene has been shown to occur in many cancer types, while the expression of PKCε was usually found to be upregulated. In this study, using immunohistochemistry, we showed that PKCε expression was lower in adjacent tissues compared with ccRCC cancer tissues, and that HepaCAM was expressed in an inversely related manner. In addition, HepaCAM was expressed strongly in adjacent noncancerous kidney tubule tissues but expression was completely lost in the most ccRCC tissues. Perhaps, this explains the ccRCC original development site, which is derived from kidney tubule tissue [21].
PKCε dysfunction has been shown to cause numerous cancers [13]. Translocation from the cytoplasm to the membrane in concert with phosphorylation of Ser-729 are regarded as the key steps in PKCε activation and cancer development [23]. To explore the relevance between HepaCAM and PKCε, we studied these proteins in the 786-0 clear cancer cell line infected with Ad-GFP-HepaCAM using western blotting. Western blot studies revealed that the cytosolic protein level increased while the plasma membrane protein level decreased without any changes to the total PKCε protein levels in HepaCAM-overexpressing cells. On the basis of these results, we speculate that HepaCAM can affect the PKCε Ser-729 phosphorylation process without changing its total protein level. Both proteins have been shown to localize to the cell membrane, which may be important in this process.
As a tumor suppressor gene, the forced expression of HepaCAM has the potential of abrogating many cancer cell type-related growth effects, which has been fully demonstrated in numerous previous studies [3, 7, 11]. HepaCAM displays a typical structure of an immunoglobulin (Ig)-like adhesion molecule, which includes two extracellular Ig-like domains, a transmembrane segment, and a cytoplasmic fragment [3]. Published reports indicate that the cytoplasmic domain of HepaCAM is essential to MCF7 cell function with regard to cell-matrix interaction and cell motility [24]. Interestingly, RACKε was associated with cell membrane sites [17]. Moreover, β1 integrin, a cell adhesion molecule similar to HepaCAM, was reported to interact with PKCε in renal cancer cells [25]. To collect more accurate data concerning potential interactions between HepaCAM and PKCε or other PKC isoforms, primary cell culture and immunoprecipitation techniques will be applied in subsequent future investigations.
PKCε is an interesting molecule that can modulate cell pathophysiologic changes in a bilateral manner. The upregulation of PKCε has been shown to have positive effects in cerebral ischemic reperfusion injury and Alzheimer’s disease [26, 27]. However, increased protein levels of PKCε have also been shown to occur in non-small cell lung tumors and leukemia [14, 28]. Genetic ablation of PKCε has also been shown to prevent prostate cancer development [12]. Its involvement in various conditions and diseases suggests that PKCε plays a role in various signaling pathways resulting in the modulation of both physiological and pathophysiological activities.
Activation of the Ras/Raf/MAPK, PI-3K/AKT, NF-κB, and Stat3 pathways by PKCε has been described previously [29–31]. In this study, we showed that PKCε was highly expressed in ccRCC tissues. We also demonstrated that PKCε was activated because central downstream proteins of PKCε that control the cell cycle, namely p-AKT, AKT, and cyclin D1, showed protein changes on western blots. Moreover, the colony formation assay confirmed the effectiveness of εV1-2 and HepaCAM through both inhibition of PKCε translocation. The combination of εV1-2 and HepaCAM was more potent at inhibiting 786-0 cell growth compared with HepaCAM alone. As a specific inhibitory peptide, εV1-2 alone, in theory, should have been enough to inhibit cell growth, however, our results demonstrated that εV1-2 only partially inhibited 786-0 cell growth. We speculate that differences in the tumor microenvironment and molecular pathways in tumor cells may explain the partial inhibition observed with εV1-2 alone. HepaCAM may act upstream of PKCε, and there may be many other pathways involved in HepaCAM control of 786-0 cell growth and development.
Taken together, our work showed that HepaCAM protein expression was decreased while PKCε expression was increased in ccRCC tissues, and there was a negative linear correlation between them. Exogenous expression of HepaCAM in 786-0 cells blocked PKCε translocation from cytosolic to plasma membrane fractions, preventing activation of PKCε, and HepaCAM, and εV1-2 exerted a synergistic anti-proliferative effect. This offers us a better understanding of HepaCAM and PKCε in ccRCC tumorigenesis, which may lead to the development of an appropriate therapeutic approach to be considered in the future.
References
Ljungberg B, Cowan NC, Hanbury DC et al (2010) EAU guidelines on renal cell carcinoma: the 2010 update. Eur Urol 58:398–406
Staehler M, Haseke N, Schoeppler G et al (2007) Modern therapeutic approaches in metastatic renal cell carcinoma. EAU-EBU Update Ser 5:26–37
Chung Moh M, Hoon Lee L, Shen S (2005) Cloning and characterization of hepaCAM, a novel Ig-like cell adhesion molecule suppressed in human hepatocellular carcinoma. J Hepatol 42:833–841
Yang S, Wu X, Luo C et al (2010) Expression and clinical significance of hepaCAM and VEGF in urothelial carcinoma. World J Urol 28:473–478
Xun C, Luo C, Wu X et al (2010) Expression of hepaCAM and its effect on proliferation of tumor cells in renal cell carcinoma. Urology 75:828–834
Moh MC, Zhang T, Lee LH et al (2008) Expression of hepaCAM is downregulated in cancers and induces senescence-like growth arrest via a p53/p21-dependent pathway in human breast cancer cells. Carcinogenesis 29:2298–2305
Wang Q, Luo C, Wu X et al (2013) HepaCAM and p-mTOR closely correlate in bladder transitional cell carcinoma and hepaCAM expression inhibits proliferation via an AMPK/mTOR dependent pathway in human bladder cancer cells. J Urol 190:1912–1918
Xu B, He Y, Wu X et al (2012) Exploration of the correlations between interferon-γ in patient serum and HepaCAM in bladder transitional cell carcinoma, and the interferon-γ mechanism inhibiting BIU-87 proliferation. J Urol 188:1346–1353
Lee LH, Moh MC, Zhang T et al (2009) The immunoglobulin-like cell adhesion molecule HepaCAM induces differentiation of human glioblastoma U373-MG cells. J Cell Biochem 107:1129–1138
He Y, Wu X, Luo C et al (2010) Functional significance of the HepaCAM gene in bladder cancer. BMC Cancer 10:83
Zhang QL, Luo CL, Wu XH et al (2011) HepaCAM induces G1 phase arrest and promotes c-Myc degradation in human renal cell carcinoma. J Cell Biochem 112:2910–2919
Hafeez BB, Zhong W, Weichert J et al (2011) Genetic ablation of PKC epsilon inhibits prostate cancer development and metastasis in transgenic mouse model of prostate adenocarcinoma. Cancer Res 71:2318–2327
Totoń E, Ignatowicz E, Skrzeczkowska K et al (2011) Protein kinase C ε as a cancer marker and target for anticancer therapy. Pharmacol Rep 63:19–29
Bae KM, Wang H, Jiang G et al (2007) Protein kinase C is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res 67:6053–6063
Akita Y (2002) Protein kinase C ε (PKC ε) its unique structure and function. J Bio chem 132:847–852
Kashiwagi K (2002) Importance of C1B domain for lipid messenger-induced targeting of protein kinase C. J Biol Chem 277:18037–18045
Csukai M, Chen CH, De Matteis MA et al (1997) The coatomer protein beta ‘-COP, a selective binding protein (RACK) for protein kinase C epsilon. J Biol Chem 272:29200–29206
Liron T, Chen LE, Khaner H et al (2007) Rational design of a selective antagonist of epsilon protein kinase C derived from the selective allosteric agonist, pseudo-RACK peptide. J Mol Cell Cardiol 42:835–841
Diouf B, Collazos A, Labesse G et al (2009) A 20-amino acid module of protein kinase C involved in translocation and selective targeting at cell-cell contacts. J Biol Chem 284:18808–18815
Edge SB, Byrd, Carducci M et al (2009) AJCC cancer staging handbook: from the AJCC cancer staging manual, vol 7. Springer, New York, pp 547–560
Störkel S, Eble JN, Adlakha K et al (1997) Classification of renal cell carcinoma. Cancer 80:987–989
Ruckhäberle E, Karn T, Denkert C et al (2013) Predictive value of sphingosine kinase 1 expression in neoadjuvant treatment of breast cancer. J Cancer Res Clin Oncol 139:1681–1689
England K, Rumsby MG (2000) Changes in protein kinase C epsilon phosphorylation status and intracellular localization as 3T3 and 3T6 fibroblasts grow to confluency and quiescence_ a role for phosphorylation at ser-729? Biochem J 352:19–26
Moh MC, Zhang C, Luo C et al (2005) Structural and functional analyses of a novel Ig-like cell adhesion molecule, HepaCAM, in the human breast carcinoma MCF7 cells. J Biol Chem 280:27366–27374
Brenner W, Benzing F, Gudejko-Thiel J et al (2004) Regulation of beta1 integrin expression by PKC epsilon in renal cancer cells. Int J Oncol 25:1157–1163
Bright R, Sun GH, Yenari MA et al (2008) ε PKC confers acute tolerance to cerebral ischemic reperfusion injury. Neurosci Lett 441:120–124
Nelson TJ, Cui C, Luo Y et al (2009) Reduction of beta-amyloid levels by novel protein kinase c (epsilon) activators. J Biol Chem 284:34514–34521
Slupsky JR, Kamiguti AS, Harris RJ et al (2007) Central role of protein kinase Cε in constitutive activation of ERK1/2 and Rac1 in the malignant cells of hairy cell leukemia. Am J Pathol 170:745–754
Basu A, Sivaprasad U (2007) Protein kinase Cε makes the life and death decision. Cell Signal 19:1633–1642
Garg R, Blando J, Perez CJ et al (2012) Activation of nuclear factor B (NF-B) in prostate cancer is mediated by protein kinase C epsilon (PKC epsilon). J Biol Chem 287:37570–37582
Aziz MH, Manoharan HT, Church DR et al (2007) Protein kinase C interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res 67:8828–8838
Acknowledgments
The study was done in the Department of Laboratory Diagnosis, Chongqing Medical University.
Conflict of interest
The authors have no conflicts of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tan, B., Tan, J., Du, H. et al. HepaCAM inhibits clear cell renal carcinoma 786-0 cell proliferation via blocking PKCε translocation from cytoplasm to plasma membrane. Mol Cell Biochem 391, 95–102 (2014). https://doi.org/10.1007/s11010-014-1991-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-014-1991-9