
Abstract
Live ischemia–reperfusion injury is associated with endoplasmic reticulum (ER) stress-induced apoptosis. Activation of peroxisome proliferator-activated receptor-α (PPARα) may inhibit hepatocyte apoptosis induced by oxidative stress and protect against liver injury. This study aimed to investigate the effects of PPARα activation, through a specific agonist, on ER stress-induced apoptosis in human liver hepatocellular carcinoma (HepG2) cells. HepG2 cells were challenged with H2O2 and treated with WY14643, a selective PPARα agonist, in the presence or absence of the PPARα antagonist of MK886. Cell viable assay (MTT) and immunostaining were used to evaluate cell viability. The level of apoptotic cell death was quantified through Annexin V/PI staining. Alanine aminotransferase, asparatate aminotransferase, and malondialdehyde levels were measured to determine the presence of cellular injury and oxidative stress. RT-PCR and Western blot analysis were used to detect mRNA and protein expression of PPARα, BiP, and CHOP. Immunofluorescence was utilized to determine the intracellular localization of CHOP. H2O2 and MK886 both reduced the viability of HepG2 cells, increased oxidative stress and apoptosis, up-regulated the BiP and CHOP expression, and induced CHOP translocation from the cytoplasm to the nucleus. Compared with cells treated with H2O2 alone, pre-administration of WY14643 increased cell viability, attenuated apoptosis, improved cell function, down-regulated BiP and CHOP expression and inhibited CHOP translocation. The effects of WY14643 were completely abolished using the MK886 antagonist. PPARα activation protects against H2O2-induced HepG2 cell apoptosis. The underlying mechanisms may be associated with its activation to suppress excessive ER stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Liver ischemia–reperfusion (LIR) injury often occurs in a number of clinical settings, such as liver resection surgery, liver transplantation, and hemorrhagic shock, and results in hepatic cell damage and death [1]. Previous studies have demonstrated that endoplasmic reticulum (ER) stress is an initiator of apoptosis during LIR injury [2–4]. LIR injury is associated with Ca2+ escape from the ER to the cytosol and this process in turn triggers the production of reactive oxygen species (ROS), leading to oxidative stress and producing a number of unfolded proteins that accumulate in the ER [5, 6]. To cope with the increase in unfolded proteins, mammalian cells generate a specific adaptive response called the unfolded protein response (UPR) [4, 7, 8]. Upon activation of the UPR, the chaperone BiP (GRP78) binds to misfolded proteins thereby activating proximal UPR transducer proteins, such as PKR-like ER-localized kinase (PERK), inositol requiring enzyme 1α (IRE1α), and activating transcription factor 6 (ATF6) [9, 10]. PERK autophosphorylation, IRE1α transautophosphorylation, and ATF6 activation all induce the expression of several genes including amino acid transporters, chaperones, and the C/EBP homologous protein/GADD153 (CHOP) [11–16]. Overexpression of CHOP leads to cell cycle arrest and/or apoptosis by decreasing Bcl-2 protein levels and translocating the Bax protein from the cytosol to the mitochondria [17]. Furthermore, induction of CHOP causes the cell to undergo apoptosis by up-regulating caspase-3 [17, 18].
PPARα is one of the three subtypes of peroxisome proliferator-activated receptors (PPARs), which are members of a set of nuclear receptors related to retinoid, steroid, and thyroid hormone receptors that are abundantly expressed in liver parenchymal cells. WY14643, a selective PPARα agonist, protects vital organs against various ischemia–reperfusion injuries to the heart, brain, kidney, and liver [19–21]. WY14643 decreases apoptosis of cardiomyocytes, preserves myocardial function, and resumes cardiac contractility after global cardiac IR under CPB [22]. Meanwhile, WY14643 up-regulates fatty aldehyde dehydrogenase (FALDH), which may contribute to the defensive system against oxidative and ER-related stress [23].
LIR injury is caused by oxidative and ER stress and leads to apoptosis. However, PPARα activation can protect against such injuries, although the possible molecular mechanisms behind this effect remain elusive. The aim of the present study is to evaluate whether PPARα activation by WY14643 protects against ER stress-induced HepG2 apoptosis and to identify a possible underlying mechanism.
Materials and methods
Cell culture and drug treatment
Human liver hepatocellular carcinoma (HepG2) cells, purchased from Shanghai Cell Bank, (Chinese Academy of Sciences, Shanghai, China) were cultured in the Dulbecco’s modified Eagle’s medium (GIBCO, Grand Island, NY, USA) and supplemented with 10 % fetal calf serum (GIBCO) at 37 °C in a humidified incubator containing an atmosphere air and 5 % CO2. For the MTT assay and PI-DAPI staining (see below), HepG2 cells were organized into 12 treatment groups as follows: negative control (no treatment); WY14643 treated cells (25 μM for 8 h dissolved in DMSO (Cayman Chemical, USA); DMSO alone (0.05 % for 8 h; mock treated); 0.1 mM, 1 mM, and 3 mΜ H2O2 (Sigma, USA) treated cells; 25 μM WY14643 (2 h pre-treatment) and 0.1 mM, 1 mM, and 3 mΜ H2O2 (for 6 h); and 0.05 % DMSO (2 h pre-treatment) and 0.1 mM, 1 mM, and 3 mΜ H2O2 (for 6 h). There were six groups in flow cytometry and immunofluorescence microscopy analysis as follows: negative control (no treatment), WY14643 treated cells [25 μM for 8 h dissolved in DMSO (Cayman Chemical, USA)], MK886 (an inhibitor of PPARα) alone (25 μM for 10 h), H2O2 alone (1 mM for 6 h), 25 μM WY14643 (2 h pre-treatment) and 1 mΜ H2O2 for 6 h, MK886 (25 μM, 2 h pre-treatment) and 25 μM WY14643 (2 h) and 1 mΜ H2O2 (6 h). For the quantification of asparatate aminotransferase (AST), alanine aminotransferase (ALT) and malondialdehyde (MDA) levels, RT-PCR and Western blotting analysis, HepG2 cells were randomized into six groups: negative control (no treatment), WY14643 treated cells (25 μM for 8 h), DMSO alone (0.05 % for 8 h; mock treated), 1 mΜ H2O2 (for 6 h), 25 μM WY14643 (2 h pre-treatment) and 1 mΜ H2O2 for 6 h, DMSO alone and 1 mΜ H2O2 for 6 h. Each experimental group above were randomized into five groups: negative control (no treatment), MK886 treated cells (25 μM for 8 h), 1 mΜ H2O2 (for 6 h), 25 μM WY14643 (2 h pre-treatment) and 1 mΜ H2O2 for 6 h, MK886 (25 μM, 2 h pre-treatment), and 25 μM WY14643 (2 h) and 1 mΜ H2O2 (6 h).
MTT assay
HepG2 cells were cultured in 96-well plates at a density of 1–5 × 105 cells/L (100 μl/well) for 24 h. 25 μM WY14643 or 0.05 % DMSO was added to the culture medium 2 h before H2O2. Then, cells were treated with various concentrations of H2O2 (0.1, 1, and 3 mM). After 6 h of H2O2, MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] solution (5 mg/mL) was added to each well (Sigma). The cells were incubated at 37 °C for 4 h. The reaction medium was carefully removed from each well, and 150 μl of DMSO was added to ensure solubilization of the crystals. The optical density values were measured using a microplate reader (Rayto, China) at 490 nm. Five replicates were applied per sample.
PI-DAPI staining
HepG2 cells were cultured in 24-well plates at a density of 1–5 × 104 cells/L (500 μl/well) and incubated with a final concentration of approximately 5 μg/ml DAPI (4′,6-diamidino-2-phenylindole) (Sigma) to the cell culture medium for 24 h. 25 μM WY14643 and 0.05 % DMSO were added to the culture medium 2 h before H2O2. Cells were then treated with various concentrations of H2O2 (0.1, 1, and 3 mM). After 6 h of H2O2 exposure, the reaction medium was carefully removed from each well, and the 500 μl of DMEM with 5 μg/ml propidium iodide (PI, Sigma) was added to the cells for 30 min. Images were captured using fluorescent microscopy (Olympus, Tokyo, Japan).
Flow cytometry
HepG2 cells (1–5 × 105 cell/well) were seeded in 6-well plates and treated with MK886, WY14643, DMSO, or H2O2. The cells were labeled with Annexin V and PI (BestBio Biosciences, China). The percent apoptosis was determined by flow cytometry and analyzed by Folwjo software. The percent of cell in early apoptosis was calculated by enumerating cells that were Annexin V-positive and PI-negative, while the percent of cells in late apoptosis was calculated by enumerating cells that were Annexin V-positive and PI-positive.
Alanine aminotransferase (ALT), asparatate aminotransferase (AST), and malondialdehyde (MDA) measurements
The supernatants of the HepG2 cells treated above mentioned treatments were harvested and the activities of AST, ALT and MDA released from cells were determined with commercially available kits. These assay kits were purchased from the Jiancheng Bioengineering Institute (Nanjing, China).
RT-PCR (reverse transcription-polymerase chain reaction) analysis
Total RNA was isolated by using Trizol Reagent (Invitrogen) according to the manufacturer’s instructions. Reverse transcription was performed with the AMV enzyme (Promega) using the manufacturer’s protocol. Amplification primers were 5′ GGT ATT GAA ACT GTG GGA GG 3′ (forward) and 5′ TTG TCT TCA GCT GTC ACT CG 3′ (reverse) for GRP8/BIP, 5′ GGA GCT GGA AGC CTG GTA TGA 3′ (forward) and 5′ TCC CTG GTC AGG CGC TCG ATT T 3′ (reverse) for GADD153/CHOP, and 5′ TCA CCA ACT GGG ACG ACA T 3′ (forward) and 5′ GCA CAG CCT GGA TAG CAA C 3′ (reverse) for β-actin.
Western blotting analysis
Cultured cells were collected and lysed in 2 % SDS sample buffer followed by SDS–PAGE. Proteins were transferred to PVDF membranes and blocked for 30 min at room temperature with phosphate buffered saline with 0.05 % Tween-20 (PBST) containing 5 % nonfat dry milk. The blots were washed three times with PBST and incubated at 4 °C overnight with either an anti-GRP78/BiP antibody (1:2000; Abcam, ab21685), an anti-GADD153/CHOP antibody (1:500; Santa Cruz Biotechnology, sc-793), or an anti-PPARα antibody (1:100; Santa Cruz Biotechnology, sc-1985). All primary antibodies were dissolved in PBST. The next day, the blots were washed three times with PBST, then incubated for 1 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies (1:2,000 dilution) (Deko Cytomation, Denmark) in PBST. Proteins were visualized using the ECL detection system (Thermo Scientific Pierce). The blots were stripped and incubated for 2 h with HRP-conjugated monoclonal mouse anti-GAPDH (1:2,000; KangCheng Biotechnology, KC-5G5) in PBST. Blots were developed using the ECL detection system. All the results shown were from three to four independent experiments.
Immunofluorescence
Cells grown on glass cover slides were washed with PBS and fixed in 10 % formalin solution containing 4 % formaldehyde for 20 min at 4 °C. Fixed cells were blocked with PBS containing 1 % bovine serum albumin and 0.1 % Triton X-100 for 30 min at room temperature then incubated with an anti-GADD153/CHOP antibody (1:50, Santa Cruz Biotechnology, sc-793), and visualized with an anti-rabbit IgG Alexa 594 secondary antibody (1:200, Molecular Probe). After the incubation with secondary antibodies, the cells were stained with 40 μg/ml DAPI for 10 min, and mounted. Microscopic analyses were performed to determine colocalization of the proteins.
Statistical analysis
All data are expressed as mean ± SD. To determine if significant differences occurred between the means of two groups, an unpaired two-tailed Student’s t test was applied. To determine if significant differences occurred among the means of several groups, a one way analysis of variance (ANOVA) was performed followed by Scheffe’s post hoc tests. A p-value of <0.05 was considered to be significant.
Results
PPARα activation increases cell viability
The changes in cell viability of HepG2 cells exposed to H2O2 were examined with the MTT assay and PI-DAPI staining. After treating cells with 0.1 mM H2O2 for 6 h, cell viability of the HepG2 cells was not altered. However, cell viability decreased from 100 to 59.6 ± 9.6 % with 1 mM H2O2 for 6 h and to 30.2 ± 3.5 % with 3 mM H2O2 for 6 h (P < 0.001). Pretreatment with 25 μM WY14643 prior to 1 mM or 3 mM H2O2 exposure significantly increased cell viability to 92.6 ± 14.2 % and 45.6 ± 4.0 % when compared to cells treated with H2O2 alone as assessed with MTT, respectively (P < 0.001) (Fig. 1a). Using the PI/DAPI staining, all cells were stained with DAPI and PI stained only dead cell. Cell viability as determined with this technique decreased to 56.1 ± 5.6 % with 1 mM H2O2 and to 32.0 ± 4.7 % with 3 mM H2O2 relative to the control group (P < 0.001) (Fig. 1b, c), while 25 μM WY14643 pretreatment significantly increased the cell viability to 96.4 ± 3.8 % and 52.8 ± 6.0 % when compared to that of control, respectively (P < 0.001) (Fig. 1b, c). However, cell viability was not altered with either WY14643 or DMSO alone as assessed by both MTT assay and PI-DAPI staining.

Cell viability. The effect of WY14643, DMSO, and H2O2 on HepG2 cell viability was measured by MTT assay (Fig. 1a) and PI-DAPI staining (Fig. 1b, c). In c, double labeling of DAPI (blue) and PI (red). In the graphs, H2O2 (black filled rounds, n = 6), DMSO + H2O2 (gray filled squares, n = 6), WY14643 + H2O2 (black filled triangles, n = 6). Data are presented as mean ± SD percentage of control (cells untreated with drugs). *P < 0.001 versus the control group (H2O2 0 mM); **P < 0.05 versus the H2O2 group at 1 and 3 mM. (Color figure online)
PPARα activation reduces cell apoptosis
To obtain additional evidence for H2O2-induced apoptosis and the protective effect of PPARα activation, cellular apoptosis was assessed through flow cytometry using PI and Annexin V-FITC staining that identifies dead and apoptotic cells, respectively [24]. Cells in the early stages of apoptosis were 5.67 ± 0.45 % (mock control), 6.24 ± 0.58 % (25 μM WY14643 alone), 25.2 ± 1.39 % (25 μM MK886 alone), 27.13 ± 2.01 % (1 mM H2O2 alone), 15.1 ± 2.01 % (25 μM WY14643 and 1 mM H2O2), 27.1 ± 2.08 % (25 μM MK886, 25 μM WY14643, and 1 mM H2O2), respectively (Fig. 2). Statistical analysis indicated that both MK886 alone and H2O2 alone induced significant apoptosis (P < 0.001). However, the pre-administration of WY14643 reduced the levels of apoptosis compared with H2O2 alone and MK886 inhibited the effect of WY14643 (P < 0.001) (Fig. 2).

Apoptosis rate. The effect of WY14643, DMSO, and H2O2 on HepG2 cell apoptosis was measured by Annexin V-fluorescein isothiocyanate/propidium iodide staining. The early apoptotic cells stained by Annexin-V-fluorescein isothiocyanate are located in the lower right quadrant. In the graphs, each column represents the mean ± SD of apoptosis rate. (n = 6 independent experiments). *P < 0.001 versus control; **P < 0.001 versus H2O2; # P < 0.001 versus WY14643 + H2O2
PPARα activation protects cell function
WY14643 treatment alone did not induce hepatocyte damage. Compared with the mock control group, ALT levels were significantly increased from 3.6 ± 0.8 to 74.7 ± 3.0 U/L in the cells treated with H2O2 alone and were also significantly increased from 5.5 ± 1.6 to 71.2 ± 2.7 U/L in the cells treated with MK886 (P < 0.001). AST levels were also increased from 7.2 ± 1.5 to 93.3 ± 6.7 U/L in the cells treated with H2O2 alone and from 10.9 ± 2.2 to 101.3 ± 5.1 U/L in the cells treated with MK886 (P < 0.001). The increases of ALT and AST induced by H2O2 were significantly attenuated to 17.5 ± 3.1 and 26.2 ± 7.8 U/L with pretreatment of WY14643 (P < 0.001) (Tables 1, 2), while the amelioration of PPARα agonist WY14643 was attenuated by the pretreatment of PPARα antagonist MK886.
PPARα activation attenuates oxidative stress
The MDA levels were increased significantly in cells treated with MK886 alone (from 3.35 ± 0.41 to 6.90 ± 0.43 μmol/L), and in cells treated with H2O2 alone (from 3.07 ± 0.43 to 7.54 ± 0.48 μmol/L) (P < 0.001). Pretreatment of WY14643 decreased the levels of MDA compared with the H2O2 treatment (to 3.18 ± 0.31 μmol/L) (P < 0.001). However, the levels of MDA increased significantly to 7.05 ± 0.27 μmol/L in cells treated with MK886, WY14643, and H2O2 compared with cells treated with WY14643 and H2O2 (P < 0.001) (Tables 3, 4).
Suppressive effects of PPARα on BiP and CHOP mRNA expression
Treatment with the PPARα agonist, WY14643, not only reduced the level of apoptosis in HepG2 cells due to H2O2, but it also inhibited H2O2-induced BiP (Bip/β-actin from 110.0 ± 7.3 to 76.7 ± 5.6 %) and CHOP (CHOP/β-actin from 42.7 ± 2.8 to 22.0 ± 4.1 %) mRNA expression (P < 0.001) (Fig. 3a, c, e). Treatment with the PPARα antagonist, MK886, suppressed the effect of WY14643 during H2O2-induced BiP and CHOP mRNA expression. Compared to cells treated with WY14643 and H2O2, BiP (Bip/β-actin from 59.5 ± 3.4 to 83.8 ± 4.6 %) and CHOP (CHOP/β-actin from 36.0 ± 3.6 to 74.6 ± 3.6 %) mRNA expression was up-regulated in cells treated with MK886, WY14643, and H2O2 (P < 0.001) (Fig. 3b, d, f). Additionally, MK886 significantly up-regulated BiP mRNA expression from 52.7 ± 3.3 to 78.3 ± 2.8 % and CHOP mRNA expression from 12.1 ± 1.1 to 66.4 ± 3.4 % compared to cells treated with only DMSO (P < 0.001) (Fig. 3).

BiP and CHOP mRNA expression. The effect of WY14643, MK886, DMSO and H2O2 on BiP, and CHOP mRNA expression in HepG2 cells. In the graphs, each column represents the mean ± SD of BIP/β-actin and CHOP/β-actin. (n = 6 independent experiments). *P < 0.001 versus control; **P < 0.001 versus H2O2; # P < 0.001 versus WY14643 + H2O2
PPARα alters H2O2 induced PPARα, BiP, and CHOP protein expression
WY14643, as a selective PPARα agonist, activated PPARα, however, H2O2 also up-regulated PPARα protein expression (PPARα/GAPDH from 62.6 ± 12.0 to 158.6 ± 14.6 % compared to mock controls) (P < 0.00) (Fig. 4a, e). MK886, as a selective PPARα antagonist, prevented both endogenous and extrinsic PPARα ligands to active PPARα (P < 0.001) (Fig. 4b, f). H2O2 increased the percentage of BiP/GAPDH from 40.7 ± 6.5 to 180.4 ± 11.0 % and the percentage of CHOP/GAPDH from 13.3 ± 4.7 to 188.2 ± 9.8 % (P < 0.001). Treatment with WY14643 produced a significant reduction in H2O2-induced BiP protein expression by 101.8 ± 13.2 % and CHOP protein expression by 67.6 ± 14.5 % (P < 0.001) (Fig. 4a, c, g). However, in contrast to the effects of WY14643, MK886 was able to inhibit the repression of BiP (from 31.2 ± 4.6 to 72.3 ± 5.8 %) and CHOP (from 17.4 ± 4.2 to 56.1 ± 4.1 %) protein expression (P < 0.001) (Fig. 4b, d, h). Moreover, when compared with mock control cells, MK886 treatment alone significantly induced BiP (28.0 ± 4.1 to 44.4 ± 5.3 %) (P = 0.001) and CHOP (3.4 ± 0.6 to 46.8 ± 6.0 %) expression (P < 0.001) (Fig. 4b, d, h).

BiP, CHOP, and PPARα protein level. The effect of WY14643, MK886, DMSO and H2O2 on BiP, CHOP and PPARα expression in HepG2 cells. In the graphs, each column represents the mean ± SD of BIP/GAPDH, CHOP/GAPDH and PPARα/GAPDH. (n = 6 independent experiments). *P < 0.001 versus control; **P < 0.001 versus H2O2; # P < 0.001 versus WY14643 + H2O2
Effect of PPARα on the intracellular localization of CHOP
In the control group, CHOP protein expression was concentrated in the cytoplasm at a very low level. H2O2-induced CHOP translocation to the nucleus, but the pretreatment of WY14643 dramatically prevented CHOP translocation from the cytoplasm to the nucleus. MK886 reversed the effect of WY14643, and caused CHOP to transfer to the nucleus (Fig. 5).

Localization of CHOP. The effect of WY14643, MK886 and H2O2 on localization of CHOP in HepG2 cells by immunofluorescence. Red fluorescence marks CHOP, blue fluorescence marks nuclei. The white arrow indicates that CHOP expressed in the cytoplasm, the green arrow refers to the localization of CHOP in the nuclei. (Color figure online)
Discussion
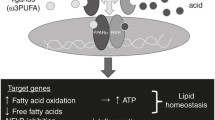
In the present study, we demonstrate that PPARα activation, through its agonist WY14643, protected hepatocytes against H2O2-induced ER stress-mediated apoptosis, due to the evidence of down-regulated BiP and CHOP expression, reduced apoptosis, and increased cellular function of this in vitro model. These protective effects were mediated by PPARα since they were abolished by MK886, a PPARα antagonist, which was demonstrated through flow cytometry analysis with Annexin V and PI, RT-PCR analysis, Western blotting analysis, and immunofluorescence. Conversely, our data also indicated that PPARα inhibition, through MK886, up-regulated BiP and CHOP mRNA and protein expression, induced apoptosis and caused liver cell damage (Fig. 6).

The protective mechanisms of PPARα activation on oxidative stress and ER stress-induced cell injury. Model showing the effect of WY14643 inhibiting the oxidative stress and ER stress-induced cell injury, in response to H2O2 through different mechanisms, which leads to the down-regulation of BiP and CHOP. As a consequence, CHOP translocation levels decrease, which allows cell survival
LIR injury is a major complication in liver surgery, particularly in liver transplantation and hepatic resection [25]. LIR effects the ability of the ER to synthesize, fold, and sort proteins and also leads to ER stress and activation of the UPR [5]. PERK, ATF6, and IRE1α are three family members of the UPR and their downstream targets such as eIF-2α, ATF4, XBP1 are activated during partial hepatectomy under IR with the induction of CHOP and BiP up-regulation and caspase-12 activation in both under steatotic and nonsteatotic conditions [26]. As reported previously [2, 4, 27], a rapid increase in CHOP is a common response of liver cells to LIR injury which may be a key pathway leading to liver cell apoptosis. There is a functional link between oxidative stress and ER stress. Increasing evidence has suggested that oxygen species (ROS) are a key mediator of LIR injury [28, 29], and accumulation of ROS in the ER increases CHOP expression and leads to ER stress and cell death [30]. Furthermore, exposure to the oxidant, tert-butyl hydroperoxide, increases oxidative stress, increases the accumulation of ROS in the ER and up-regulates expression of BiP and CHOP in human retinal pigment epithelium cells [31]. In line with a previous study [32], using a established H2O2 induced ER stress mediated apoptosis model, as reported here, our data show that oxidative stress induced by H2O2 contributes to ER stress through the up-regulation of BiP and CHOP, accompanied with a remarkable increase of MDA, ALT, and AST release in response to H2O2 challenge associated with CHOP localization in the nucleus (Fig. 5).
Activation of PPARα provides protection against organ injury resulting from ischemia reperfusion. For example, a PPARα agonist WY14643 significantly reduced the severity of liver pathology [33], and also provided a protective effect on dietary steatohepatitis, which results from the action of ROS on accumulated lipids and excessive formation of lipoperoxides in the livers of mice [34]. In the brain, WY14643 prevents ROS production in hippocampal neurons and protects from β-amyloid induced neurodegeneration [21]. Our previous study [20] also showed that WY14643 administration before the onset of liver ischemia significantly protects liver against LIR injury in rats. Its effect is probably associated with the enhancement of anti-oxidative capacities, inhibition of inflammation and enhanced expression of antioxidant enzymes such as SOD, decreased MDA levels in the liver, and decreased levels of TNF-α, IL-1β and MPO in the plasma. Pretreatment of WY14643 exerts a protective effect against hypoxia/reoxygenation injury in primary hepatocyte cultures by activating PPARα, attenuating oxidative stress and these effects occur in a dose-dependent manner [35]. However, the mechanism by which PPARα is activated with WY14643 and provides a protective effect is still unknown. The current data reported here show that WY14643 activates PPARα, and completely down-regulated the expression of BiP and CHOP at both the transcriptional and translational level, while both BiP and CHOP mediate the development of ER stress to trigger cell apoptosis. Furthermore, we found that WY14643 blocked H2O2-induced translocation of CHOP from cytoplasm to nucleus. Administration of WY14643 activated PPARα and increased PPARα expression, while MK886, a selective antagonist of PPARα, inhibited the activation of PPARα. Ultimately, MK886 triggers cells to become apoptotic, however, WY14643 activates PPARα mediating the anti-apoptotic effect to protect cells [36].
In summary, our data indicate that WY14643 protected HepG2 cells against H2O2-induced apoptosis and MK886 reduced cell viability and increased apoptosis. Furthermore, MK886 abolished the anti-apoptotic effect of WY14643 on H2O2-induced apoptosis via suppressing activation of PPARα. Interestingly, we also found that H2O2 stimulated the up-regulation of endogenous PPARα in HepG2 cells. The mechanisms for this observation are unknown but warrant further study.
Our study is not without limitations: first, a liver cancer cell line was used. Although they behave and respond to drug treatments, this may be different to normal primary cells. Secondly, a relatively high concentrations of H2O2 used in our experiments and are likely irrelevant clinically. Third, MK886, as a noncompetitive PPARα antagonist, may not completely block WY14643 on the activation of PPARα. Further studies, therefore, are required to determine whether PPARα activation would suppress excessive ER stress and its associated changes as shown in our study in a clinically relevant model of LIR injury.
References
Yang L-Q, Tao K-M, Liu Y-T, Cheung C-W, Irwin MG, Wong GTC, Lv H, Song J-G, Wu F-X, Yu W-F (2011) Remifentanil preconditioning reduces hepatic ischemia–reperfusion injury in rats via inducible nitric oxide synthase expression. Anesthesiology 114:1036–1047
Bailly-Maitre B, Fondevila C, Kaldas F, Droin N, Luciano F, Ricci J-E, Croxton R, Krajewska M, Zapata JM, Kupiec-Weglinski JW, Farmer D, Reed JC (2006) Cytoprotective gene bi-1 is required for intrinsic protection from endoplasmic reticulum stress and ischemia–reperfusion injury. Proc Natl Acad Sci USA 103:2809–2814
Mosbah IB, Alfany-Fernández I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, Rodés J, Brenner C, Roselló-Catafau J, Peralta C (2010) Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia–reperfusion. Cell Death Dis 1:e52
Mosbah IB, Zaouali MA, Martel C, Bjaoui M, Abdennebi HB, Hotter G, Brenner C, Rosello-Catafau J (2012) IGL-1 solution reduces endoplasmic reticulum stress and apoptosis in rat liver transplantation. Cell Death Dis 3:e279. doi:10.1038/cddis.2012.12
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565. doi:10.1038/nrm1150
Cheng F, Zhong X, Lu Y, Wang X, Song W, Guo S, Liu D, Wang Q (2012) Refined QingKaiLing protects MCAO mice from endoplasmic reticulum stress-induced apoptosis with a broad time window. Evid Based Complement Altern Med 2012:567872. doi:10.1155/2012/567872
Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, Hotamisligil GS (2011) Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473:528–531
Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS (2006) Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 313:1137–1140. doi:10.1126/science.1128294
Malhi H, Kaufman RJ (2011) Endoplasmic reticulum stress in liver disease. J Hepatol 54:795–809
Fuest M, Willim K, MacNelly S, Fellner N, Resch GP, Blum HE, Hasselblatt P (2012) The transcription factor c-Jun protects against sustained hepatic endoplasmic reticulum stress thereby promoting hepatocyte survival. Hepatology 55:408–418
Harding HP, Zhang Y, Ron D (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397:271–274. doi:10.1038/16729
Teske BF, Wek SA, Bunpo P, Cundiff JK, McClintick JN, Anthony TG, Wek RC (2011) The eIF2 kinase PERK and the integrated stress response facilitate activation of ATF6 during endoplasmic reticulum stress. Mol Biol Cell 22:4390–4405. doi:10.1091/mbc.E11-06-0510
Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 6:1099–1108
Luo S, Baumeister P, Yang S, Abcouwer SF, Lee AS (2003) Induction of Grp78/BiP by translational block: activation of the Grp78 promoter by ATF4 through and upstream ATF/CRE site independent of the endoplasmic reticulum stress elements. J Biol Chem 278:37375–37385. doi:10.1074/jbc.M303619200
Richardson CE, Kooistra T, Kim DH (2010) An essential role for XBP-1 in host protection against immune activation in C. elegans. Nature 463:1092–1095. doi:10.1038/nature08762
Ye J, Rawson RB, Komuro R, Chen X, Dave UP, Prywes R, Brown MS, Goldstein JL (2000) ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell 6:1355–1364
Oyadomari S, Mori M (2004) Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11:381–389. doi:10.1038/sj.cdd.4401373
Xu C, Bailly-Maitre B, Reed JC (2005) Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest 115:2656–2664. doi:10.1172/jci26373
Li J, Zúñiga J, Cancino M, Medina F, Varela P, Vargas R, Tapia G, Videla LA, Fernández V (2011) N-3 PUFA supplementation triggers PPAR-α activation and PPAR-α/NF-κB Interaction: anti-inflammatory implications in liver ischemia–reperfusion injury. PLoS ONE 6:e28502
Xu S-Q (2008) Effects of Wy14643 on hepatic ischemia reperfusion injury in rats. World J Gastroenterol 14:6936–6942. doi:10.3748/wjg.14.6936
Santos MJ, Quintanilla RA, Toro A, Grandy R, Dinamarca MC, Godoy JA, Inestrosa NC (2005) Peroxisomal proliferation protects from beta-amyloid neurodegeneration. J Biol Chem 280:41057–41068. doi:10.1074/jbc.M505160200
Yeh CH, Chen TP, Lee CH, Wu YC, Lin YM, Lin PJ (2006) Cardiomyocytic apoptosis following global cardiac ischemia and reperfusion can be attenuated by peroxisome proliferator-activated receptor alpha but not gamma activators. Shock 26:262–270. doi:10.1097/01.shk.0000225863.56714.96
Ashibe B, Motojima K (2009) Fatty aldehyde dehydrogenase is up-regulated by polyunsaturated fatty acid via peroxisome proliferator-activated receptor α and suppresses polyunsaturated fatty acid-induced endoplasmic reticulum stress. FEBS J 276:6956–6970
Vanags DM, Porn-Ares MI, Coppola S, Burgess DH, Orrenius S (1996) Protease involvement in fodrin cleavage and phosphatidylserine exposure in apoptosis. J Biol Chem 271:31075–31085
de Rougemont O, Lehmann K, Clavien P-A (2009) Preconditioning, organ preservation, and postconditioning to prevent ischemia–reperfusion injury to the liver. Liver Transplant 15:1172–1182. doi:10.1002/lt.21876
Peralta C, Brenner C (2011) Endoplasmic reticulum stress inhibition enhances liver tolerance to ischemia/reperfusion. Curr Med Chem 18:2016–2024
Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, Eckhoff DE, Contreras JL (2005) Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery 138:342–351. doi:10.1016/j.surg.2005.04.019
Zhou G, Dada LA, Wu M, Kelly A, Trejo H, Zhou Q, Varga J, Sznajder JI (2009) Hypoxia-induced alveolar epithelial-mesenchymal transition requires mitochondrial ROS and hypoxia-inducible factor 1. Am J Physiol Lung Cell Mol Physiol 297:L1120–L1130. doi:10.1152/ajplung.00007.2009
Song X, Zhang N, XU H, Cao L, Zhang H (2012) Combined preconditioning and postconditioning provides synergistic protection against liver ischemic reperfusion injury. Int J Biol Sci 8:707–718. doi:10.7150/ijbs.4231
Moon D-O, Park S-Y, Choi YH, Ahn JS, Kim G-Y (2011) Guggulsterone sensitizes hepatoma cells to TRAIL-induced apoptosis through the induction of CHOP-dependent DR5: involvement of ROS-dependent ER-stress. Biochem Pharmacol 82:1641–1650. doi:10.1016/j.bcp.2011.08.019
He S, Yaung J, Kim YH, Barron E, Ryan SJ, Hinton DR (2008) Endoplasmic reticulum stress induced by oxidative stress in retinal pigment epithelial cells. Graefes Arch Clin Exp Ophthalmol 246:677–683. doi:10.1007/s00417-008-0770-2
Pan C, Giraldo GS, Prentice H, Wu J-Y (2010) Taurine protection of PC12 cells against endoplasmic reticulum stress induced by oxidative stress. J Biomed Sci 17:S17. doi:10.1186/1423-0127-17-s1-s17
Schrader M, Fahimi HD (2006) Peroxisomes and oxidative stress. Biochim Biophys Acta 1763:1755–1766. doi:10.1016/j.bbamcr.2006.09.006
Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I (2003) Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 38:123–132. doi:10.1053/jhep.2003.50307
Chen K, Li YH, Xu SQ, Hu SH, Zhang L (2012) Protective effects of peroxisome proliferator-activated receptor-alpha agonist, Wy14643, on hypoxia/reoxygenation injury in primary rat hepatocytes. PPAR Res 2012:547980. doi:10.1155/2012/547980
Schuster M, Zouboulis CC, Ochsendorf F, Müller J, Thaçi D, Bernd A, Kaufmann R, Kippenberger S (2011) Peroxisome proliferator-activated receptor activators protect sebocytes from apoptosis: a new treatment modality for acne? Br J Dermatol 164:182–186
Acknowledgments
This work was financially supported by National Nature Science Foundation (81171031). The authors are grateful to the Key Laboratory of Gene Resource Utilization for Genetic Diseases of Educational Ministry, Anhui Medical University, Hefei, China, for providing instructions of technology and material support. The authors also thank to Dr. Daqing Ma, M.D., Ph.D., Imperial College London for his critical comment during manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Li-kui Wang: co-first author.
Rights and permissions
About this article
Cite this article
Tang, Wx., Wang, Lk., Wang, Yq. et al. Peroxisome proliferator-activated receptor-α activation protects against endoplasmic reticulum stress-induced HepG2 cell apoptosis. Mol Cell Biochem 385, 179–190 (2014). https://doi.org/10.1007/s11010-013-1826-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-013-1826-0