
Abstract
Protein kinases are important regulators in biologic processes. Aberrant expression of protein kinases often causes diseases including cancer. In the present study, we found that the serine-arginine protein kinase 1 (SRPK1) might be involved in hepatocellular carcinoma (HCC) proliferation from a kinome screen using a loss-of-function approach. In clinical samples, SRPK1 was frequently up-regulated in HCCs as compared with adjacent non-tumor tissues at both mRNA and protein levels. Functional studies indicated that overexpression of wild-type SRPK1 promoted HCC cell proliferation, while forced expression of the kinase-dead mutant of SRPK1 or RNA interference against SRPK1 suppressed cell growth and malignancy as measured in soft agar assay. The kinase-dead mutant of SRPK1 also inhibited subcutaneous xenografts’ growth of HCC cells in nude mice. Furthermore, western bolt analysis showed overexpression of wild-type SRPK1 enhanced Akt phosphorylation and knockdown of SRPK1 by RNA interference attenuated Akt phosphorylation induced by epidermal growth factor. Meanwhile, overexpression of wild-type SRPK1 also induced a concurrent increase in the total tyrosine phosphorylation of phosphotidylinositol-3 kinase p110α subunit, indicating a functional link between SRPK1 and PI3K/Akt signaling. Our findings suggest that SRPK1 plays an oncogenic role and could be a potential therapeutic target in HCC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
At molecular or cellular levels, factors that cause cancer include gene mutations [1, 2], genomic instability [3, 4], and deregulation in mechanisms that maintain a balance between cellular growth, proliferation, differentiation, and apoptosis [5]. Research into these molecular and cellular mechanisms could deepen our understanding of cancer and at the same time provide valuable insights into designing therapeutic interventions.
Phosphorylation is a well-known mechanism mediating protein functions and signaling transduction; it is estimated that over one third of total proteins inside cells are phosphorylated and thus regulated by protein kinases [6]. In the meantime, protein kinase itself is under strict regulation. Mutation and alterations in expression are both possible causes that disturb the fine-tuned networking of protein kinases, which often have pathological consequences including cancer. Such examples include, among numerous others, CDCs and CDKs, the kinases that are involved in cell cycle control and chromosome duplication, and also DAPKs and MAPKs, the kinases that actively participate in proliferation, differentiation, and apoptosis [3, 7–9]. It is safe to say that virtually most cases of protein kinase deregulations in normal cell functions can find their representation in tumor transformation or metastasis.
Serine-arginine protein kinase 1 (SRPK1) is a highly conserved protein in eukaryotic organisms; it contains a Pkc superfamily kinase domain and a spacer region that divides the kinase domain by half. SRPK1 mainly phosphorylates specific amino acids of proteins rich in serine/arginine repeats known as RS-domain proteins. RS-domain proteins are proteins that participate in precursor mRNA translation, processing and splicing, chromatin reconstruction, cell cycle progression, and remodeling of cellular structures. Deletion of the spacer region alters SRPK1 cellular residence from primarily localized in cytoplasm to almost exclusively in nuclei [10].
It seems that roles of SRPK1 in different types of cancers are conflicted. Reports identified that elevation of SRPK1 expression level correlated with risks of breast, colon, and pancreatic cancers [11, 12]. In a different study in retinoblastoma (RB), SRPK1 expression level was down-regulated, and RB patients with low level of SRPK1 had poor response toward cis-platin treatment, suggesting that the endogenous level of SRPK1 functions to suppress tumor transformation and progression [13]. The precise roles and mechanisms of SRPK1 in cancer development remain to be elucidated. It might be expected that SRPK1 plays diverse roles in cancer progression. Mutations in WT1, the Wilms’ tumor suppressor gene, cause abnormal gonadogenesis, renal failure, and Wilms’ tumors [14]. In WT1 mutant cells, SRPK1-mediated hyperphosphorylation of the oncogenic RNA-binding protein SRSF1 regulated splicing of VEGF and rendered WT1 mutant cells proangiogenic, thus promoting tumor progression [14].
Studies to date have provided a characterization on molecular and cellular traits of SRPK1, the roles of which in hepatocellular carcinoma (HCC) are anticipated. HCC, the major histologic subtype of liver cancer, is one of the most frequent malignancies in the world and the third leading cause of cancer-related death [15]. According to one study, in the process of infection, SRPK1 mediated the phosphorylation of HBV core protein, which had been shown to be a prerequisite for pregenomic RNA encapsidation in viral capsid [16]. Since chronic HBV infection is an important risk factor in liver cancer, studying SRPK1 will not only help understand the carcinogenesis of HCC but also provide valuable insights into HCCs associated with chronic HBV infection.
In the present study, we found that SRPK1 mRNA and protein expressions were significantly elevated in HCCs compared to the adjacent non-cancerous tissues in patients of liver cancer. Further investigation identified SRPK1 as a potential oncoprotein that affected various aspects of HCC, including proliferation, malignancy, and tumor growth of in vitro HCC xenograft. In addition, we also found a functional link between SRPK1 and PI3K/Akt signaling.
Materials and methods
Tissue specimens and cell lines
All specimens were harvested from 27 HCC patients, with their informed consent, who underwent resection of the primary tumor at the department of Wuxi People’s Hospital. The paired adjacent non-tumor liver samples were from the same patients and 2 cm away from the edge of tumor regions. The histopathologic diagnosis was based on the criteria of the World Health Organization, and over 50 % of tumor cells in HCC tumoral samples were observed in the morphology. Thirteen liver tumor-derived cell lines and the fetal liver-derived cell line L02 were used in this study. All cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (Fisher Scientific) supplemented with 1 % penicillin/streptomycin and 10 % fetal bovine serum (Life Technologies) in a 5 % CO2-humidified chamber. Protocols involving animals and human samples had been approved by the ethics committee of the Chinese National Human Genome Center at Shanghai and Wuxi People’s Hospital, as was previously described [17].
Quantitative real-time polymerase chain reaction
Total RNA was extracted from the tissue samples using TRIZOL reagent (invitrogen) and was reverse transcribed into cDNA using the M-MLV reverse transcriptase kit (Promega). The following primers were used to amplify human SRPK1: forward, CACGGCATGCATGGCCTTTGA; reverse, CGGCGGCAGTGGCTCTCTTC. Quantitative real-time PCR was performed with the Takara PCR thermal cycler dice detection system and SYBR green dye (Takara) in paired HCC specimens. The mean Ct value for the β-actin gene was subtracted from the mean Ct value for SRPK1 for each sample, using the following formula: SRPK1ΔCt = (mean SRPK1Ct − mean actinCt), SRPK1ΔΔCt = (SRPK1ΔCt_HCC − SRPK1ΔCt_non-HCC). The fold change (2−SRPK1ΔΔCt) of the SRPK1 expression level relative to the β-actin expression level was calculated for each HCC sample. Statistical significance was defined as p < 0.05 in paired, two-tailed student’s t test.
Transfection of plasmids and siRNAs against SRPK1
Plasmids or siRNAs were transfected into cells using lipofectamine 2,000 (invitrogen) according to the manufacturer’s instructions. Two sequences of siRNAs against SRPK1 were synthesized from Shanghai GenePharma Co. Sense sequences for the two siRNAs against SRPK1 are as follows: #1: GUGCAGCAGAAAUUAAUU; #2: GAUCAUCAAAUCCAAUU. Efficacies of the siRNAs were verified by western blotting detection of cellular SRPK1.
Cell proliferation and soft agar colony formation assays
Cell proliferation was measured using the cell counting kit-8 (Dojindo Laboratories) according to the manufacturer’s instructions. For soft agar assay, 1.5 ml of culture medium containing 1 % agarose was added into each well of six-well plates; the plates were set aside for 15 min to allow the agarose to solidify. Forty-eight h after transfection, cells were trypsinized; around 10,000 cells were resuspended into 1.5 ml of culture medium containing 0.5 % agarose and the mixtures were added on top of the 1 % agarose base layer. After solidification of the top layer, cells were fed 1–2 times per week with 1.5 ml culture medium. Plates were incubated at 37° in a humidified incubator for 3 weeks and stained with 0.5 ml of 0.005 % crystal violet for >1 h.
Plasmid constructs and adenoviruses generation
The full length wild-type SRPK1 (SRPK1-WT) cDNA was amplified from mRNA of L02 cells by RT-PCR. The kinase-dead mutant of SRPK1 (SRPK1-KD) was generated by mutating the critical catalytic lysine of amino acid 109 into alanine. SRPK1-WT and SRPK1-KD were inserted into pcDNA3.1(-)A-myc/his expression vector (invitrogen). Adenovirus expressing SRPK1-WT or SRPK1-KD was generated using the AdEasyTM XL adenoviral vector system (stratagene) according to the manufacturer’s instructions. All constructs were verified for sequence correctness by direct sequencing.
Immunoprecipitation and western blotting
Immunoprecipitation and western blotting were performed as previously described [18]. Cell lysis buffer was supplemented with 1 mM Na3VO4 and proteinase inhibitors cocktail (Sigma). Mouse anti-SRPK1 antibody was from BD Biosciences; mouse anti-myc antibody was from Santa Cruz; Rabbit anti-phospho-tyrosine antibody (PY100), rabbit anti-PI3K p110α, rabbit anti-Akt, and rabbit anti-phospho-Akt (473) antibodies were all from cell signaling technology.
Subcutaneous xenograft of HCC cells
SK-Hep1 or WRL-68 cells were infected with adenoviruses expressing control vector or SRPK1-KD. A total of 2 × 106 infected cells were subcutaneously injected into each flank of the nude mice. Procedures were done as previously described [19]. Growth curves were plotted based on mean tumor volume within each experimental group at the indicated time point. Tumor dimensions were measured twice every week using a digital caliper. Tumor volume was calculated by the formula V = 0.5 × (larger diameter) × (smaller diameter)2. Tumor growth was observed for at least 3 weeks. The nude mice were sacrificed at the end of experiment, and tumors were excised and weighed. Statistical significance was defined as p < 0.05 in paired, two-tailed student’s t test.
Results
SRPK1 expression level was frequently elevated in HCC
To identify kinases involved in HCC proliferation, we performed a screen using a loss-of-function approach of depleting 636 kinases individually in four HCC cell lines (Huh-7, Hep3B, MHCC-H, and MHCC-L). Those kinases with inhibition of cell anchorage-dependant growth were identified (unpublished data). Among them, we focused on the SRPK1, of which the function in HCC remains unclear.
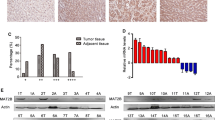
We first tested the relative expression of SRPK1 in HCC specimens through real-time polymerase chain reaction. As shown in Fig. 1a, SRPK1 mRNA levels were aberrantly elevated in tumors compared to their paired adjacent non-malignant liver tissues. Among the samples, 17 of 27 (63.0 %) displayed at least a 1.5-fold increase in HCC tissues compared to non-HCC tissues (Fig. 1b). Meanwhile, SRPK1 expression was also analyzed by western blotting. Consistent with the mRNA level, the protein expression of SRPK1 was significantly up-regulated in 58.3 % (7/12) of HCCs (Fig. 1c). In addition, we also explored the expression profile of SRPK1 in various liver cancer cell lines. As shown in Fig. 1d, all liver cancer cell lines indicated abundant expression of SRPK1, indicating its involvement in HCC carcinogenesis. These data suggest that SRPK1 is frequently elevated in HCC and may act as a positive factor toward HCC.

SRPK1 expression is elevated in HCC specimens and cell lines. a Transcript level of SRPK1 was measured in HCC specimens and corresponding adjacent non-cancerous liver tissues by quantitative RT-PCR. β-actin was used as an internal loading control. Each plotting dot represents the expression level of a given HCC specimen or adjacent liver tissue. Lines represent the median with interquartile range of −ΔCt value. N non-HCC tissues, C HCC tissues. p value was calculated by student’s t test. b Relative expression fold of SRPK1 mRNA for 27 paired HCC samples used in (a), each pair consisted of HCC tissue (C) and non-HCC tissue (N) from the same patient. c Protein expression level of SRPK1 was measured in HCC specimens and corresponding adjacent non-cancerous liver tissues by western blotting. d Protein expression profile of SRPK1 in HCC cells lines was assessed by western blotting as described in the Materials and methods section
SRPK1 affects cellular proliferation
SRPK1 plays its role mainly by phosphorylating specific amino acids of proteins rich in serine/arginine repeats known as RS-domain proteins. To produce an effect that mimics a pharmaceutical inhibitor, we generated a kinase-dead mutant of SRPK1 by mutating the critical catalytic lysine to alanine (K109A); this mutant was then designated as SRPK1-KD [20, 21]. Next, we investigated the effects of wild-type SRPK1 and kinase-dead SRPK1 on cellular proliferation in L02 and WRL-68 cells. The results demonstrated that wild-type SRPK1 enhanced, while kinase-dead SRPK1 inhibited the cellular proliferation in these two HCC cell lines (Fig. 2a). On the other hand, we also explored the effect of SRPK1 silencing by RNAi on cellular proliferation in YY-8103 and WRL-68 cells. Silencing of SRPK1 had a markedly inhibitory effect on cellular proliferation as shown in Fig. 2b. These collective data suggest that SRPK1 is involved in the regulation of HCC cell proliferation.

Wild-type SRPK1 promotes, while kinase-dead SRPK1 or silencing SRPK1 inhibits cellular proliferation. a Wild-type or kinase-dead SRPK1 was transfected into L02 or WRL-68 cells; after transfection, cells were seeded into 96-well plate with each well containing 3,000 cells; for the following 72 h, cell density was monitored once every 24 h by staining cells with CCK-8 and then measuring the absorbance rate at a wavelength of 450 nm. The ectopic expression of wild-type SRPK1 and kinase-dead SRPK1 was confirmed by western blotting in L02 and WRL-68 cells. b Silencing SRPK1 by siRNA against SRPK1 inhibits cellular proliferation in YY-8103 and WRL-68 cells using a similar method as described in a. Western blot images showed the silencing effect of SRPK1 RNAi
Kinase-dead SRPK1 or siRNA-mediated knockdown of SRPK1 suppresses HCC malignancy
Maintaining malignant growth is a critical step of carcinogenesis, and anchorage-independent growth in soft agar is an important characteristic of tumor malignancy [22]. To examine the effect of SRPK1 on malignancy of HCC cells, we performed soft agar colony formation assays in three HCC cell lines SK-Hep1, YY-8103, and WRL-68, which have the ability to grow in soft agar. The cells transfected with SRPK1-KD or siRNAs against SRPK1 decreased their ability to form colonies in soft agar compared to those transfected with vector control or siR-NC (Fig. 3a, b). These results indicated that SRPK1 inactivation or knockdown suppressed the malignant growth of HCC cells. However, we did not observe any effect of wild-type SRPK1 overexpression in this growth manner (Fig. 3c). One possible reason is that the cell lines we used already have relatively high level expression of SRPK1. The efficacies of SRPK1 overexpression and siRNA transfection were confirmed by western blotting (Supplementary Fig. 1). These observations suggest that SRPK1 is critical for maintaining the malignant growth of HCC cells and also implies that inhibiting SRPK1 kinase activity might be an effective route to suppressing malignancy of HCC tumors.

SRPK1 siRNA or ectopic expression of SRPK1-KD suppresses anchorage-independent growth of HCC cells in soft agar. a Ectopic expression of SRPK1-KD suppressed anchorage-independent growth of HCC cells in soft agar. SK-Hep1 was stably transfected with SRPK1-KD. YY-8103 or WRL-68 cells were transiently transfected with SRPK1-KD. b SRPK1 siRNA suppressed anchorage-independent growth of HCC cells in soft agar. WRL-68 or YY-8103 cells were transfected with SRPK1 siRNA. c SRPK1 overexpression has no obvious effect on anchorage-independent growth of SK-Hep-1 and WRL-68 cells. SK-Hep-1 was stably transfected with SRPK1-WT (wild-type), and WRL-68 cells were transiently transfected with SRPK1-WT
SRPK1 is functionally linked with PI3K/Akt signaling
To explore the potential mechanism through which SRPK1 contributes to HCC, we tested the effect of overexpression of SRPK1 on a number of factors and signaling molecules that had been known to play important roles in cellular proliferation. These molecules included MAPKs, Src, PI3K, and Akt. In our case, as shown in Fig. 4a, overexpression of SRPK1 induced an increase in the phosphorylated level of Akt, indicating SRPK1 overexpression increased the activity of Akt, but without much alteration in total Akt level. On the contrary, knockdown of SRPK1 attenuated EGF-induced phosphorylation of Akt (Fig. 4b). To explore the mechanism by which overexpression of SRPK1 activates Akt, we tested whether SRPK1 has an effect on PI3K, the upstream of Akt signaling. Total tyrosine phosphorylation of p110 subunits was immunoprecipitated from Huh-7 or YY-8103 cells transfected with SRPK1-WT or SRPK1-KD by a pan-phospho-tyrosine antibody. The results showed that the tyrosine phosphorylation level of p110α subunit was increased when wild-type SRPK1 was overexpressed (Fig. 4c, d), implying that PI3K signaling is probably affected by SRPK1.

Ectopic SRPK1-WT expression induced Akt phosphorylation, and SRPK1 may be functionally linked with PI3K. a Ectopic SRPK1-WT expression induced phosphorylation of Akt. b siRNA knockdown of SRPK1 suppressed epidermal growth factor-induced Akt phosphorylation. YY-8103 cells were transfected with wild-type SRPK1 or siRNAs against SRPK1(#1 and #2 represent two different siRNA sequences); 48 h after transfection, cells were serum starved for 3 h and treated with EGF (10 ng/ml) for 15 min as indicated. Cells were then lysed and phospho-Akt level was measured by western blotting. c, d Ectopic expression of SRPK1-WT, but not of SRPK-KD induced an increase in the level of tyrosine phosphorylation of PI3K p110α subunit. Huh-7 or YY-8103 Cells were transfected with SRPK1-WT or SRPK1-KD; 48 h after transfection, cells were lysed and immunoprecipitation was performed with anti-phospho-tyrosine antibody (pY-100). The presence of p110α in the immunoprecipitates was detected by western blotting with antibody to p110α
Kinase-dead SRPK1 inhibits tumor growth of subcutaneous xenograft of HCC cells in nude mice
In the previous experiments, we have demonstrated that the kinase-dead mutant of SRPK1 inhibited cellular proliferation and anchorage-independent growth of HCC cells. To further characterize the effect of inhibition of SRPK1 kinase activity on HCC, we generated an adenovirus that expresses the kinase-dead mutant of SRPK1. After infection with this adenovirus, the HCC cells were subcutaneously injected into nude mice to assess the effect of SRPK1 on tumorigenicity. Tumor sizes were monitored twice a week for 4 weeks. As shown in Fig. 5, infection of HCC cells with adenoviruses expressing kinase-dead SRPK1 significantly suppressed the growth potential of the subcutaneous xenograft of HCC cells. The expression of SRPK1-KD was confirmed by western blotting (Fig. 5c). We also tested the effect of wild-type SRPK1 on the growth potential of subcutaneous xenograft of HCC cells; no significant difference was observed between vector control and wild-type SRPK1 groups (Supplementary Fig. 2).

SRPK1-KD suppresses tumor growth of subcutaneous xenograft of HCC cells in nude mice. SK-Hep1 or WRL-68 cells were infected with adenoviruses expressing control vector or SRPK1-KD. A total of 2 × 106 infected cells were subcutaneously injected into each flank of nude mice. Tumor dimensions were measured twice every week using a digital caliper. Tumor growth was observed for at least 3 weeks. Nude mice were sacrificed at the end of experiment, and tumors were excised and weighed. a SK-Hep1 cells, b WRL-68 cells. p < 0.05 in paired student’s t test. c The viral expression of SRPK1-KD in SK-Hep1 and WRL-68 cells was confirmed by western blotting
Discussion
SRPK1 has been reported in a number of human cancers, though its roles in different cancers are debated. However, there is no report on the role of SRPK1 in HCC to date. In the present study, we identified SRPK1 as an oncoprotein in HCC for the first time. Our clinical data showed that SRPK1 mRNA and protein levels are frequently higher in HCCs compared to the adjacent non-cancerous tissues, in agreement with those expression patterns of breast, colon, and pancreatic cancers. Thus, SRPK1 may play similar roles in these cancer developments. Previous structural studies have shown that SRPK1 is resilient to inactivation by mutation in its activation loop and remains active despite extensive mutation to the activation segment [23], suggesting that changes in its expression level are major aspects in SRPK1-related pathogenesis under in vivo circumstances.
We further showed that inhibition of endogenous SRPK1 either by siRNA or kinase-dead mutant suppressed many aspects of HCC malignancies including proliferation, anchorage-independent growth, and growth potential of in vitro HCC xenografts in nude mice. Surprisingly, however, in our soft agar assays and subcutaneous xenograft experiments, we did not observe an increase in growth potential of HCC cells overexpressing wild-type SRPK1, possibly due to the fact that HCC cell lines in our study already had elevated expression of SRPK1. Our data suggest that endogenous SRPK1 kinase activity might be crucial in maintaining the malignancies of HCCs rather than promoting transformation of normal liver cells into HCCs.
Aberrant upregulation of epidermal growth factor (EGF) signaling has been shown to participate in a number of tumors, in which PI3K/Akt plays a major part [24, 25]. To explore potential molecular mechanisms by which SRPK1 functions in promoting cellular proliferation, the effect of overexpression of SRPK1 on PI3K/Akt signaling was studied by western blotting. Our data demonstrated that upon EGF stimulation, overexpression of SRPK1 induced Akt phosphorylation, but without much alteration in total Akt level, while knockdown of SRPK1 did the opposite, indicating that SRPK1 is required for EGF-mediated AKT activation. Usually, stimulation-induced phosphorylation of p85 releases p110 from association with and inhibition by p85 subunit, and the released p110 then mediates phosphorylation and activation of Akt or other downstream targets [26]. In the present study, p110α subunit was found to be present in the anti-phospho-tyrosine antibody immunoprecipitates, indicating a concurrent increase in the tyrosine phosphoryaltion of p110α. Our data suggested that SRPK1 is functionally linked with PI3K/Akt signaling and may act as upstream of Akt. Of note, a recent report demonstrated that EGF can activate an AKT-SRPK axis to control alternative splicing [27]. In this setting, activated Akt induces SRPK1 autophosphorylation and this leads to enhanced SRPK1 nuclear translocation and downstream splicing factors phosphorylation, suggesting that SRPK1 may be located downstream of Akt. A possible feedback loop between Akt and SRPK1 is depicted in Fig. 6. The detailed mechanism by which SRPK1 induces Akt activation and whether autophosphorylated SRPK1 could mediate this process deserve to be further investigated.

A schematic diagram of the contribution of SRPK1 to HCC via activation of Akt. The process in which SRPK1 mediates EGF signaling transduction to Akt is unknown. A feedback loop may exist between Akt and SRPK1 as indicated
Our results demonstrated that aberrant elevation in SRPK1 expression contributed to malignancy of HCC through a possible mechanism involving PI3K/Akt signaling. Since chronic HBV infection is also an important risk factor in liver cancer and HBV replication involves SRPK1 kinase activity of its host hepatocytes in the process of infection, SRPK1 might be a promising target of pharmaceutical intervention, especially for HCCs with chronic HBV infection.
References
Berger MF, Hodis E, Heffernan TP et al (2012) Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 485:502–506. doi:10.1038/nature11071
Hsu IC, Metcalf RA, Sun T et al (1991) Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature 350:427–428. doi:10.1038/350427a0
Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B (2004) Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene 23:4014–4022. doi:10.1038/sj.onc.1207505
Schwartzentruber J, Korshunov A, Liu XY et al (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231. doi:10.1038/nature10833
Ho L, Stojanovski A, Whetstone H, Wei QX, Mau E, Wunder JS, Alman B (2009) Gli2 and p53 cooperate to regulate IGFBP-3- mediated chondrocyte apoptosis in the progression from benign to malignant cartilage tumors. Cancer Cell 16:126–136. doi:10.1016/j.ccr.2009.05.013
Cohen P (2000) The regulation of protein function by multisite phosphorylation–a 25 year update. Trends Biochem Sci 25:596–601
Duvvuri U, Shiwarski DJ, Xiao D et al (2012) TMEM16A induces MAPK and contributes directly to tumorigenesis and cancer progression. Cancer Res 72:3270–3281. doi:10.1158/0008-5472.CAN-12-0475-T
Xiao D, Herman-Antosiewicz A, Antosiewicz J, Xiao H, Brisson M, Lazo JS, Singh SV (2005) Diallyl trisulfide-induced G(2)-M phase cell cycle arrest in human prostate cancer cells is caused by reactive oxygen species-dependent destruction and hyperphosphorylation of Cdc 25 °C. Oncogene 24:6256–6268. doi:10.1038/sj.onc.1208759
Zhang D, Vuocolo S, Masciullo V, Sava T, Giordano A, Soprano DR, Soprano KJ (2001) Cell cycle genes as targets of retinoid induced ovarian tumor cell growth suppression. Oncogene 20:7935–7944. doi:10.1038/sj.onc.1204971
Ding JH, Zhong XY, Hagopian JC, Cruz MM, Ghosh G, Feramisco J, Adams JA, Fu XD (2006) Regulated cellular partitioning of SR protein-specific kinases in mammalian cells. Mol Biol Cell 17:876–885. doi:10.1091/mbc.E05-10-0963
Hayes GM, Carrigan PE, Beck AM, Miller LJ (2006) Targeting the RNA splicing machinery as a novel treatment strategy for pancreatic carcinoma. Cancer Res 66:3819–3827. doi:10.1158/0008-5472.CAN-05-4065
Hayes GM, Carrigan PE, Miller LJ (2007) Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res 67:2072–2080. doi:10.1158/0008-5472.CAN-06-2969
Krishnakumar S, Mohan A, Kandalam M et al (2008) SRPK1: a cisplatin sensitive protein expressed in retinoblastoma. Pediatr Blood Cancer 50:402–406. doi:10.1002/pbc.21088
Amin EM, Oltean S, Hua J et al (2011) WT1 mutants reveal SRPK1 to be a downstream angiogenesis target by altering VEGF splicing. Cancer Cell 20:768–780. doi:10.1016/j.ccr.2011.10.016
Llovet JM, Burroughs A, Bruix J (2003) Hepatocellular carcinoma. Lancet 362:1907–1917. doi:10.1016/S0140-6736(03)14964-1
Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A, Cotten M (2002) Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol 76:8124–8137
Deng Q, Wang Q, Zong WY, Zheng DL, Wen YX, Wang KS, Teng XM, Zhang X, Huang J, Han ZG (2010) E2F8 contributes to human hepatocellular carcinoma via regulating cell proliferation. Cancer Res 70:782–791. doi:10.1158/0008-5472.CAN-09-3082
Zhou B, Zhuang J, Gu D, Wang H, Cebotaru L, Guggino WB, Cai H (2010) WNK4 enhances the degradation of NCC through a sortilin-mediated lysosomal pathway. J Am Soc Nephrol 21:82–92. doi:10.1681/ASN.2008121275
Huang J, Zheng DL, Qin FS, Cheng N, Chen H, Wan BB, Wang YP, Xiao HS, Han ZG (2010) Genetic and epigenetic silencing of SCARA5 may contribute to human hepatocellular carcinoma by activating FAK signaling. J Clin Invest 120:223–241. doi:10.1172/JCI38012
Jang SW, Yang SJ, Ehlen A, Dong S, Khoury H, Chen J, Persson JL, Ye K (2008) Serine/arginine protein-specific kinase 2 promotes leukemia cell proliferation by phosphorylating acinus and regulating cyclin A1. Cancer Res 68:4559–4570. doi:10.1158/0008-5472.CAN-08-0021
Yeakley JM, Tronchère H, Olesen J, Dyck JA, Wang HY, Fu XD (1999) Phosphorylation regulates in vivo interaction and molecular targeting of serine/arginine-rich pre-mRNA splicing factors. J Cell Biol 145(3):447–455 May 3
Gatza CE, Holtzhausen A, Kirkbride KC, Morton A, Gatza ML, Datto MB, Blobe GC (2011) Type III TGF-beta receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia 13:758–770
Ghosh G, Adams JA (2011) Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J 278:587–597. doi:10.1111/j.1742-4658.2010.07992.x
El Sheikh SS, Domin J, Abel P, Stamp G, el Lalani N (2004) Phosphorylation of both EGFR and ErbB2 is a reliable predictor of prostate cancer cell proliferation in response to EGF. Neoplasia 6:846–853. doi:10.1593/neo.04379
Martin-Orozco RM, Almaraz-Pro C, Rodriguez-Ubreva FJ, Cortes MA, Ropero S, Colomer R, Lopez-Ruiz P, Colas B (2007) EGF prevents the neuroendocrine differentiation of LNCaP cells induced by serum deprivation: the modulator role of PI3K/Akt. Neoplasia 9:614–624
Neri LM, Borgatti P, Capitani S, Martelli AM (2002) The nuclear phosphoinositide 3-kinase/AKT pathway: a new second messenger system. Biochim Biophys Acta 1584:73–80
Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, Fu XD (2012) The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell Aug 10 47(3):422–433
Acknowledgments
We thank Drs. Xiao Xu and Rui-fang Liu for helpful discussions and technical assistance. This work was supported by grants from the Chinese National Key Program on Basic Research (973 Program) (2010CB529204 and 2010CB529206), the Chinese National Key Projects for Infectious Disease(2012ZX10002012-008), and the Chinese Postdoctoral Science Foundation Award (20110490764).
Disclosure of Potential Conflict of interest
The authors declare that no conflicts of interest exist.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bo Zhou and Yandong Li contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Zhou, B., Li, Y., Deng, Q. et al. SRPK1 contributes to malignancy of hepatocellular carcinoma through a possible mechanism involving PI3K/Akt. Mol Cell Biochem 379, 191–199 (2013). https://doi.org/10.1007/s11010-013-1641-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-013-1641-7