
Abstract
Chronic alcohol consumption causes numerous biochemical and biophysical changes in the central nervous system, in which mitochondria is the primary organelle affected. In the present study, we hypothesized that alcohol alters the mitochondrial membrane properties and leads to mitochondrial dysfunction via mitochondrial reactive oxygen species (mROS) and reactive nitrogen species (RNS). Alcohol-induced hypoxia further enhances these effects. Administration of alcohol to rats significantly increased the mitochondrial lipid peroxidation and protein oxidation with decreased SOD2 mRNA and protein expression was decreased, while nitric oxide (NO) levels and expression of iNOS and nNOS in brain cortex were increased. In addition, alcohol augmented HIF-1α mRNA and protein expression in the brain cortex. Results from this study showed that alcohol administration to rats decreased mitochondrial complex I, III, IV activities, Na+/K+-ATPase activity and cardiolipin content with increased anisotropic value. Cardiolipin regulates numerous enzyme activities, especially those related to oxidative phosphorylation and coupled respiration. In the present study, decreased cardiolipin could be ascribed to ROS/RNS-induced damage. In conclusion, alcohol-induced ROS/RNS is responsible for the altered mitochondrial membrane properties, and alcohol-induced hypoxia further enhance these alterations, which ultimately leads to mitochondrial dysfunction.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chronic alcohol induced neurotoxicity is associated with changes in mitochondria function [1–4]. Mitochondria are the target of ethanol intoxication mainly due to the increased production of reactive oxygen species/reactive nitrogen species (ROS/RNS) during ethanol metabolism [5]. Previous reports revealed that chronic alcohol consumption inhibits complexes I and III of the mitochondrial electron transport chain and increases ROS production in liver [6, 7]. Studies also revealed that ethanol consumption increases nitric oxide (NO) level and may lead to toxicity by peroxynitrite, a potent oxidant which produces nitration of tyrosine and inactivation of many biologically important proteins and enzymes [8]. Hence, alcohol-induced oxidative/nitrosative stress leads to the imbalance between levels of ROS/RNS and the antioxidant defense capacity, resulting in damage to critical cellular macromolecules such as lipids, DNA, and proteins [9]. Chronic alcohol consumption affects mitochondrial function, including oxygen homeostasis and leads to a hypoxic condition at high blood alcohol levels [10, 11]. In addition to the oxidative and nitrosative stress, hypoxia has also been suggested as a key factor capable of both initiating and sustaining the mechanisms of pathogenesis. Hypoxia-inducible factors (HIFs) are the master regulators of oxygen homeostasis. Cells respond to low O2 availability by initiating a series of adaptive responses through transcriptional activation and stabilization of HIF-1α [12]. HIF-1α activates nitric oxide synthase (NOS) expression and on the other hand NO inhibits HIF-1α DNA binding activity and HIF-1α expression in hypoxic cells [13]. The modulation of NO by the hypoxic response is believed to have wide pathophysiological significance in mitochondria [14]. Further, mitochondrial damage plays a significant role in this process. Cytochrome c oxidase contains an oxygen binding site and is the principal target for the interaction of NO in regulating several mitochondrial functions including formation of ROS/RNS [15].
Alcohol affects the physical properties and functions of membranes, which leads to alterations in ion channels and receptors and disruption of membrane lipids [16]. Mitochondrial respiration is an important source of ROS production and hence a potential contributor to oxidative injury to membranes. Phospholipids play multiple roles in biologic membranes. Cardiolipin (1,3-diphosphatidyl-sn-glycerol) (CL) is a complex mitochondrial-specific phospholipid that regulates numerous enzyme activities, especially those related to oxidative phosphorylation and coupled respiration [17]. CL binds complexes I, III, IV, and V and stabilizes the super complexes (I/III/IV and II/III/IV), demonstrating an absolute requirement of CL for catalytic activity of these enzyme complexes [18]. CL is particularly susceptible to oxidative attack due to its high content of unsaturated fatty acids. Alterations in the content and/or composition of CL have been shown to be responsible for mitochondrial dysfunction in a variety of pathological settings [19].
The activity of most membrane-bound enzymes is regulated by the physicochemical state of their membrane lipid environment [20]. Studies have shown that alcohol-induced generation of free radicals affects membrane-linked enzyme activity through the modification of membrane fluidity in erythrocyte membranes [16]. Na+/K+-ATPase is a membrane protein critically important for the maintenance of the ion gradients required for cell homeostasis. Studies also revealed that hypoxia mediates degradation of Na+/K+-ATPase via mitochondrial ROS [21]. The intoxicating effect of alcohol on the brain can be attributed to its modification of membrane fluidity and alterations of membrane proteins, receptors, and ion channels. The mechanisms by which these effects are brought about are not fully known. Therefore, we hypothesized that alcohol-induced ROS/RNS is responsible for the altered mitochondrial membrane properties, and alcohol-induced hypoxia further enhances these alterations, which ultimately leads to mitochondrial dysfunction. The pattern of results presented here help to elucidate the molecular mechanism of ROS/RNS-induced damage to mitochondrial membrane constituents, which contributes to mitochondrial dysfunction.
Materials and methods
Chemicals
The chemicals used in the present study were procured from Sigma Chemical Co. (St. Louis, MO, USA) and SISCO Research Laboratories (Mumbai, India). Ethanol used for administration to rats was obtained by re-distillation.
Animals and experimental design
Two-month-old male albino Wistar rats weighing about 120–140 g were procured from Sri Venkateswara Enterprises, Bangalore, India. They were fed with commercial pellet diet (Hindustan Lever Ltd, Mumbai, India) and tap water ad libitum. The animals were divided into alcohol and control groups of eight rats in each group. Control rats received glucose instead of alcohol (i.e., caloric equivalent to alcohol), alcohol-treated rats received 20 % (v/v) alcohol in water at a dosage of 5 g/kg body weight/day. Both groups received treatment for 60 days by forced oral administration with the help of a stomach tube. Food and water intake of all the animals was recorded daily and the weight of rats was followed on alternate days. At the end of the experimental period, the rats in each group were fasted overnight and then euthanized by cervical dislocation. Brain tissues were collected and processed immediately for further analysis.
Mitochondria TBARS, protein carbonyls, nitrite and nitrate analyses
Mitochondria were isolated from brain cortex as described earlier [22] and the purity of the mitochondria were assessed by analyzing the mitochondrial complex activities. Thiobarbituric acid reactive species (TBARS) were measured by the formation of malondialdehyde [23]. Protein carbonyls were determined as described previously [24]. Nitrite/nitrate levels were determined as described previously [25].
Measurement of mitochondrial complex I, II, III, and IV activity
Complex I (NADH dehydrogenase) activity was measured spectrophotometrically using potassium ferricyanide as electron acceptor and the activity was expressed as NADH units per minute per milligram protein [26]. Complex II, succinate dehydrogenase enzyme was assayed using 2,6-dichlorophenolindophenol (DCPIP) as electron acceptor and the activity was expressed as units per minute per milligram protein [27]. Complex III, cytochrome bc 1 complex (ubiquinol–ferricytochrome c oxido-reductase) of the mitochondria was measured as described previously [28]. The activity was measured in the presence and absence of antimycin A and expressed as nanomoles of cytochrome c reduced per minute per milligram protein. Complex IV, cytochrome c oxidase activity was assayed as described earlier [29]. Activity was expressed as nmol of cytochrome c oxidized per minute per milligram protein.
Measurement of mitochondria ATPases
Na+/K+-ATPase activity was determined as described earlier [30]. The activities were indirectly measured by estimating the phosphorus liberated after the incubation of mitochondria in a reaction mixture containing the substrate ATP with the co-substrate elements at 37 °C for 15 min. The reactions were arrested by adding 1.0 ml of 10 % trichloroacetic acid (TCA). The phosphorus content from the TCA supernatants was then determined [31].
Lipid extraction and determination of cardiolipin analysis
Lipids were extracted from equal amount of control and alcohol-treated mitochondria protein samples as described previously [32]. CL was separated on silica gel H (Merck) by two dimensional thin layer chromatography with chloroform–methanol–aqueous ammonia 65:35:5 (v/v) as the first solvent and chloroform–acetone–methanol–acetic acid–water 50:20:10:10:5 (v/v) as the second solvent. The fractions were located using iodine vapors and were scraped from the plate; the CL was measured as inorganic phosphorus after digestion with perchloric acid [31]. Values were expressed as microgram of CL per mg protein.
Membrane fluidity studies using DPH
The quantitative measurement of membrane fluidity was performed by the fluorescence polarization technique described previously with 1,6-diphenyl-1,3,5-hexatriene (DPH) as fluorescence probe [33]. Membranes (50 μg protein) were suspended in 50 mmol/L DPH solubilized in tetrahydrofuran, and incubated at 37 °C for 30 min. Fluorescence polarization was determined using a Hitachi fluorescence spectrophotometer (Hitachi, Japan) equipped with rotating polarizing filters with samples held at 25 °C. Samples were excited at 360 nm and the emission intensity was read at 435 nm. Polarization (P) and fluorescence anisotropy (γ) were calculated by the equation. P = I VV − I VH·G /I VV + I VH·G, where I VV and I VH are the intensities measured parallel and perpendicular to the vertical axis of the excitation beam, and G is the correction factor. I VH /I HH· γ is calculated using the formula γ = 2P/(3 − P). Protein concentration was estimated by the method of Lowry et al. [34].
Western blotting
Rat brain cortex was homogenized in radio immunoprecipitation assay (RIPA) buffer containing 50 mM Tris–HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.4 % deoxy cholate, 1 % NP-40 containing protease inhibitors including 1 mM phenylmethylsulfonylfluoride (PMSF), and phosphatase inhibitors including 10 mM β-glycerophosphate, 10 mM NaF, 0.3 mM Na3Vo4 and 0.3 mM aprotinin. The lysate was sonicated and centrifuged at 14,000×g for 15 min at 4 °C. Cellular protein was mixed with SDS sample buffer subjected to electrophoresis and transferred onto PVDF membranes. After blocking the PVDF membrane in non-fat dry milk (5 %) in Tris buffered saline (TBS) for 1 h at room temperature, membranes were incubated for overnight with iNOS (Cat# N32020; Transduction laboratories, Lexington, USA), nNOS (Cat# N31020; Transduction laboratories, Lexington, USA), HIF-1α (Cat# NB100-123; Novus Biologicals, Littleton, CO, USA), SOD2 (Cat# 06-984; Millipore, Billerica, MA, USA) and Tubulin (Sigma, St Louis, MO, USA) primary antibodies. Then, the membranes were incubated with secondary antibodies (anti-rabbit and anti-mouse IgG conjugated to HRP) for 1 h at room temperature. Before and after incubation with secondary antibodies, membranes were washed with TBS containing 0.1 % Tween-20. Membranes were analyzed quantitatively by means of image J software (NIH).
Analysis of HIF-1α and SOD2 mRNA expression by RT-PCR
Real-time reverse transcriptase (RT)-PCR was performed using a MiniOpticon system (Bio-Rad) with SYBR GreenER using qRT-PCR kit (Bio-Rad) as previously described [35]. Briefly, RNA was extracted from cerebral cortex using TRIZOL and a 1 μg aliquot was reverse transcribed using Superscript III (Bio-Rad). Primer sequences for real-time RT-PCR amplification are shown in Table 1. Relative mRNA level (R) was calculated based on the comparative threshold (C T) method by the formula \( R = 2^{{ - \Updelta C_{T} }} \), where ∆C T is the difference between the threshold cycle of the given target mRNA in controls and alcohol-treated rat tissues. The C T value was taken as a fractional cycle number at which the emitted fluorescence of the sample passes a fixed threshold above the baseline and the values were normalized to an internal standard (18S rRNA). Purity and specificity of all products were confirmed by omitting the template and by performing a standard melting curve analysis.
Statistical analysis
Student’s t test was used for statistical significance between controls and alcohol administered rats. Values are mean ± SD of all the parameters, A *p < 0.05 is statistically significant from controls.
Results
Effect of alcohol on mitochondrial TBARS, protein carbonyls, and SOD2 mRNA and protein expression
In the present study, we investigated the effect of alcohol on oxidative stress status by measuring TBARS, protein carbonyls, and mitochondrial localized SOD2 mRNA and protein expression. The data presented in Fig. 1a–d reveal the oxidative status in both controls and alcohol-treated rats. Alcohol administration to rats significantly increased mitochondrial TBARS (Fig. 1a) and protein carbonyl (Fig. 1b) levels. Further, we noticed significantly decreased mitochondrial specific antioxidant, SOD2 mRNA (Fig. 1c) and protein (Fig. 1d) expression in alcohol treated rats.

Effect of alcohol-induced oxidative stress in the brain cortex. TBARS (a) and protein carbonyls (b) in mitochondria and SOD2 mRNA (c) and protein (d) levels were measured. Total protein samples were prepared using RIPA buffer. Brain cortex lysate protein (10 μg/well) was used for western blot analyses. Data are presented as mean ± SD from six independent experiments, and expressed as the percentage of the control. The control values for TBARS and protein carbonyls are 1.36 ± 0.06 nmol MDA/mg protein and 1.76 ± 0.14 nmol/mg protein, respectively. A p < 0.05 is statistically significant from controls. Asterisk indicates statistical significance compared with controls
Influence of alcohol on brain cortex nitrite/nitrate levels and iNOS and nNOS protein and mRNA expression
Data presented in Fig. 2a–c show the effect of alcohol administration on nitrosative stress. We analyzed NO production by measuring nitrate and nitrite levels as well as NOS iso-forms, iNOS and nNOS, protein expression in brain cortex. Administration of alcohol significantly increased nitrate and nitrite levels as well as iNOS and nNOS protein expression.

Effect of alcohol on nitrite/nitrate (NO) level, iNOS and nNOS protein levels in the brain cortex. (a) NO levels in the brain cortex were measured by using Gries reagent as described in “Materials and methods”. The control value of NO is 13.1 ± 1.2 nmol/mg protein. Brain cortex lysate protein (50 μg/well) was used for western blot analyses. Immunoreactive bands of (b) iNOS, (c) nNOS and tubulin protein, results are expressed as mean ± SD of five independent experiments, and presented as μmol/mg cellular protein (for NO), or as ratio of nNOS or iNOS to that of tubulin-immunoreactive bands. A p < 0.05 is statistically significant from controls. Asterisk indicates statistical significance compared with controls
Influence of alcohol on protein and mRNA expression of HIF-1α
To study the effect of alcohol on HIF-1α, we analyzed HIF-1α protein expression by western blotting and mRNA expression by quantitative real-time RT-PCR. Administration of alcohol significantly increased both protein and mRNA expression of HIF-1α compared to controls (Fig. 3).

Effect of alcohol on HIF-1α mRNA and protein levels in the brain cortex. (a) HIF-1α mRNA and (b) HIF-1α protein levels in brain cortex homogenates were analyzed. Total protein samples were prepared using RIPA buffer and subjected to western blot analysis. The total RNA was isolated using Trizol reagent as described in “Materials and methods”. Data are presented as mean ± SD from five independent experiments, and were normalized to a house-keeping protein or gene. A p < 0.05 is statistically significant from controls. Asterisk indicates statistical significance compared with controls
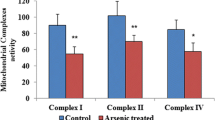
Influence of alcohol on mitochondrial complex activities
To study mitochondrial function, we determined mitochondrial complex activities in control and alcohol administered rat brain mitochondria. The activities of complex I (Fig. 4a), complex III (Fig. 4c), and complex IV (Fig. 4d) of mitochondria were significantly decreased, but there was no significant effect on complex II (Fig. 4b) in brain mitochondria of ethanol-fed rats compared to controls.

Effect of alcohol on mitochondrial complex activities. Mitochondrial complex activities were measured as described in “Materials and methods”. (a) Complex I, (b) complex II, (c) complex III, and (d) complex IV. Data are presented as mean ± SD from six independent experiments, and expressed as the percentage of the control. The control values for complex I, complex II, complex III, and complex IV are 2,763 ± 34, 183.7 ± 6.67, 1,125 ± 54 and 1,848 ± 26, respectively. Asterisk indicates statistical significant from controls. *p < 0.05; ns not significant
Influence of alcohol on mitochondrial membrane properties
We studied the membrane properties by measuring membrane fluidity, Na+/K+-ATPase activity and inner mitochondrial localized phospholipid, CL. Membrane fluidity was studied using DPH, a fluorescent probe, partitions selectively into the membrane core constituted by phospholipid fatty acid acyl chains and provides essential information concerning the fluidity in membranes. DPH studies revealed that alcohol administration resulted in a considerable increase in anisotropy (γ) values indicating decreased fluidity evidenced from a decrease in DPH polarization in mitochondrial membranes (Fig. 5b). Our results also showed that alcohol administration significantly decreased CL content (Fig. 5a) and Na+/K+-ATPases activity (Fig. 5c) in mitochondria.

Effect of alcohol on mitochondrial membrane properties. Mitochondrial membrane properties were studied by measuring cardiolipin content, membrane fluidity, and Na+/K+-ATPase activity. Results represent in (a) cardiolipin, (b) anisotropic value, and (c) Na+/K+-ATPase activity in controls and alcohol administered rats. Data are presented as mean ± SD from six independent experiments and were expressed as the percentage of the control. The control values for cardiolipin, anisotropic (γ) value and Na+/K+-ATPase activity are 18.5 ± 0.62 μg/mg protein, 0.318 ± 0.01 U/mg protein and 5.51 ± 0.1 μmol pi liberated/h/mg protein, respectively. Asterisk indicates statistical significant from controls. *p < 0.05, **p < 0.01
Discussion
Chronic intake of alcohol alters cell membrane fluidity by complex neurochemical adaptive changes and neurochemical alterations. Increased oxidative/nitrosative stress is likely responsible for these changes. The balance between ROS production and antioxidant defenses determines the oxidative stress status. Alcohol can stimulate lipid peroxidation by the formation of free radicals or by exhausting antioxidant substances, leading to oxidative stress in the brain [36–39]. Consequences of this stress include modification of cellular proteins, DNA, and lipids, particularly phospholipids, with high degree of unsaturation. Highly reactive products of lipid peroxidation can react and modify the structure and function of membrane proteins and enzymes [40]. ROS production is largely counteracted by an intricate antioxidant defense system that includes SOD2 in mitochondria. The decreased SOD2 is due to the increased ROS production in alcohol-treated rats. In the present study, increased lipid peroxidation and protein oxidation may be due to an increase in ROS and a decrease in antioxidant levels. Over production of NO might also be playing an important role in alcohol induced events [41]. Both O2 •− and NO• generated from mitochondria combine and form peroxynitrite (ONOO•−). In vivo ONOO− generation represents a crucial pathogenic mechanism in many diseased conditions [42]. Free radicals generated from alcohol metabolism can directly damage cell membranes by peroxidation of membrane polyunsaturated fatty acids. Results from the study clearly indicated that increased membrane lipid peroxidation and protein oxidation in mitochondria could be due to increased free radicals generation by alcohol and it is in agreement with earlier studies [9, 43].
Following alcohol consumption, oxygen uptake is increased during alcohol metabolism leading to hypoxic conditions. Adaptation to hypoxia is a key for cell survival. HIF-1α is the major transcription factor controlling cellular adaptation to hypoxia. During hypoxia, HIF-1α activates many target genes, including those involved in NO synthesis, erythropoiesis, angiogenesis, glycolysis, and cell proliferation [12]. Chronic alcohol administration increases the expression of HIF-1α and is in agreement with previous reports showing increased HIF-1α expression in liver after ethanol treatment [7, 44]. Our lab also has shown that alcohol administration increases the expression of HIF-1α and its target genes, erythropoietin (EPO), and vascular endothelial growth factor (VEGF) in the blood of human chronic alcoholics (data not shown). Adaptation to alcohol-induced hypoxia is achieved by upregulating HIF-1α, which regulates the efficiency of vascular oxygen transport and energy supply, inducing several genes. The generation of mitochondrial reactive oxygen species (mROS) during hypoxia has been proposed as part of an oxygen sensing pathway for the hypoxic stabilization of HIF-1α [45]. In the present study, increased iNOS and nNOS mRNA and protein expression has been observed after alcohol treatment in the brain cortex. This increase may involve a general cellular stress response, leading to increased gene transcription, or direct activation of transcription through binding of HIF-1α.
Phospholipids play multiple roles in biologic membranes. Phospholipids may undergo oxidative damage in their acyl chains due to oxygen radical attack. This leads to changes in structural characteristics and dynamics of the lipid bilayer with functional impairment in membrane fluidity, ion permeability, membranous enzyme activity, and cell signaling [46]. CL is a structurally unique dimeric phospholipid localized in the inner mitochondrial membrane, and it is required for optimal mitochondrial function. In addition to its role in maintaining membrane potential and architecture, it provides essential structural and functional support to several proteins involved in mitochondrial bioenergetics [47]. A loss of CL content, alterations in its acyl chain composition, and/or CL peroxidation have been associated with mitochondrial dysfunction. In the present study, a loss in the CL content could be due to the decrease in CL synthase activity or CL peroxidation due to ROS attack in alcohol-administered rats. The activity of respiratory enzyme complexes I and III and their linked activities are directly related to CL content [48]. Decreased mitochondrial complex I, III, and IV activities could be due to decreased CL content. A decrease in cellular oxygen tension will exacerbate the impaired mitochondrial electron flow caused by alcohol by decreasing the delivery of O2 to the mitochondria [7]. Under low oxygen conditions, complex III produces O2 •− and complex IV produces NO•, both combine and produce ONOO•− [14, 49, 50]. It was reported that complex III contains tightly bound CL which is essential for the catalytic function. Besides the impairment of complex III activity due to the ROS-induced CL, oxidative damage may increase the electron leak from the electron transport chain, generating more superoxide radicals and perpetuating a cycle of oxygen radical-induced damage, which ultimately leads to mitochondrial dysfunction in alcohol-treated rats.
Phospholipids have been shown to be essential for providing the proper polarity and special arrangement necessary for optimal activity of membrane-bound enzymes. In the present study, decreased CL in mitochondrial membrane showed altered phospholipid milieu of the membrane affecting the lipid packing, structure, and organization of the membrane and the subsequent changes in the functioning and orientation of several membrane proteins. Fluidity of the lipid environment influences physiologically important membrane-bound enzymes such as Na+/K+-ATPases, important for the maintenance of the Na+/K+ ion gradients, which are required for cell homeostasis. Previous studies have shown that Na+/K+-ATPase is sensitive to changes in membrane fluidity [51, 52]. In the present study, the observed increase in anisotropic value in alcohol-treated animals suggested decreased fluidity in mitochondrial membranes. Alcohol-induced generation of free radicals also affects Na+/K+-ATPase enzyme activity through the modification of membrane fluidity because the activity of most membrane-bound enzymes is regulated by the physicochemical state of their membrane lipid environment. Earlier studies suggest that ROS are generated at the mitochondria, specifically at complex III, playing a key role in the degradation of the Na+ pump during hypoxia [21]. Thus, cells undergoing hypoxia are exquisitely prone to oxidative stress. Results suggest that alcohol-induced distortions in the mitochondrial membrane might lead to interference with respiratory chain functioning, loss of membrane potential, and conformational changes in proteins with attendant changes in the Na+/K+-ATPase activity. The rate of peroxidation also affects the fluidity of the membrane. In the present study, decrease in the activity of Na+/K+-ATPase in alcohol-administered rats occurs by two mechanisms: (i) alcohol-induced hypoxia could degrade Na+/K+-ATPase, thereby altering membrane properties via mitochondrial ROS, (ii) oxidative stress could induce the modification of membrane phospholipids and proteins, which are responsible for the inhibition of ATPase activity.
In conclusion, it is pertinent to note from the study that alcohol-induced oxidative damage to CL negatively impacts the biochemical function of the mitochondrial membranes by altering membrane fluidity, ion permeability, and function of mitochondrial electron transport chain components resulting in reduced mitochondrial oxidative phosphorylation. Alcohol-induced hypoxia and nitrosative stress further enhances these effects. The molecular mechanisms underlying the role of alcohol and consequential effects on membrane properties as well as key signaling pathways which mediate the effects of alcohol on mitochondrial function need further in-depth study.
References
Harper C, Matsumoto I (2005) Ethanol and brain damage. Curr Opin Pharmacol 5:73–78
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Almansa I, Fernandez A, García-Ruiz C, Muriach M, Barcia JM, Miranda M, Fernández-Checa JC, Romero FJ (2009) Brain mitochondrial alterations after chronic alcohol consumption. J Physiol Biochem 65:305–312
Reddy VD, Padmavathi P, Kavitha G, Gopi S, Varadacharyulu N (2011) Emblica officinalis ameliorates alcohol-induced brain mitochondrial dysfunction in rats. J Med Food 14:62–68
Bailey SM (2003) A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic Res. 37:585–596
Venkatraman A, Shiva S, Wigley A, Ulasova E, Shhieng D, Bailey SM, Darley-Usmar VM (2004) The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatol 40:565–573
Zelickson BR, Benavides GA, Johnson MS, Chacko BK, Venkatraman A, Landar A, Betancourt AM, Bailey SM, Darley-Usmar VM (2011) Nitric oxide and hypoxia exacerbate alcohol-induced mitochondrial dysfunction in hepatocytes. Biochim Biophys Acta 1807:1573–1582
Deng X, Deitrich RA (2007) Ethanol metabolism and effects: nitric oxide and its interaction. Curr Clin Pharmacol 2:145–153
Haorah J, Ramirez SH, Floreani N, Gorantla S, Morsey B, Persidsky Y (2008) Mechanism of alcohol-induced oxidative stress and neuronal injury. Free Radic Biol Med 45:1542–1550
Young TA, Bailey SM, Van Horn CG, Cunningham CC (2006) Chronic ethanol consumption decreases mitochondrial and glycolytic production of ATP in liver. Alcohol 41:254–260
Wang SM, Wu R (2009) The double danger of ethanol and hypoxia: their effects on a hepatoma cell line. Int J Clin Exp Pathol 2:182–189
Semenza GL (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148:399–408
Taylor CT, Moncada S (2010) Nitric oxide, cytochrome c oxidase, and the cellular response to hypoxia. Arterioscler Thromb Vasc Biol 30:643–647
Ball KA, Nelson AW, Foster DG, Poyton RO (2012) Nitric oxide produced by cytochrome c oxidase helps stabilize HIF-1α in hypoxic mammalian cells. Biochem Biophys Res Commun 420:727–732
Poyton RO, Ball KA, Castello PR (2009) Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab 20:332–340
Reddy VD, Padmavathi P, Paramahamsa M, Varadacharyulu N (2009) Modulatory role of Emblica officinalis against alcohol induced biochemical and biophysical changes in rat erythrocyte membranes. Food Chem Toxicol 47:1958–1963
Paradies G, Petrosillo G, Paradies V, Ruggiero FM (2009) Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease. Cell Calcium 45:643–650
Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H (2003) Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278:52873–52880
Chicco AJ, Sparagna GC (2007) Role of cardiolipin alterations in mitochondrial dysfunction and disease. Am J Physiol Cell Physiol 292:C33–C44
Muriel P, Sandoval G (2000) Nitric oxide and peroxynitrite anion modulate liver plasma membrane fluidity and Na+/K+-ATPase activity. Nitric Oxide 4:333–342
Comellas AP, Dada LA, Lecuona E, Pesce LM, Chandel NS, Quesada N, Budinger GR, Strous GJ, Ciechanover A, Sznajder JI (2006) Hypoxia-mediated degradation of Na, K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ Res 98:1314–1322
Brustovetsky W, Dubinsky JM (2000) Dual responses of CNS mitochondria to elevated calcium. J Neurosci 20:103–113
Ohkawa H, Ohishi N, Yagi K (1979) Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem 95:351–358
Reznick AZ, Packer L (1994) Oxidative damage to proteins: spectroscopic method for carbonyl assay. Methods Enzymol 233:357–363
Sastry KHV, Moudgal RP, Mohan J, Tyagi JS, Rao GS (2002) Spectrophotometric determination of serum nitrite and nitrate by copper–cadmium alloy. Anal Biochem 306:79–82
King TE, Robert HL (1967) Preparations and properties of soluble NADH dehydrogenases from cardiac muscle. In: Estabrook RW, Pullman ME (eds) Methods in enzymology, vol 10. Academic Press Inc., New York, pp 275–294
King TE (1967) Preparation of succinate dehydrogenase and reconstitution of succinate oxidase. In: Estabrook RW, Pullman ME (eds) Methods enzymol, vol 10. Academic Press Inc., New York, pp 322–331
Krahenbuhl S, Talos C, Wiesmann U, Hoppel CL (1994) Development and evaluation of a spectrophotometric assay for complex III in isolated mitochondria, tissues and fibroblasts from rats and humans. Clin Chim Acta 230:177–187
Cooperstein SJ, Lazarow A (1951) A microspectrophotometric method for the determination of cytochrome oxidase. J Biol Chem 189:665–670
Ismail B, Edelman IS (1985) Assay of Na+, K+-ATPase. Biochem Pharmacol 34:2685–2689
Fiske CH, Subbarow Y (1925) The colorimetric determination of inorganic phosphorus. J Biol Chem 66:375–404
Folch J, Ascoli I, Lees M, Meath JA, Le Baron FN (1951) Preparation of lipid extracts from brain tissue. J Biol Chem 191:833–841
Padmavathi P, Reddy VD, Kavitha G, Paramahamsa M, Varadacharyulu N (2010) Chronic cigarette smoking alters erythrocyte membrane lipid composition and properties in male human volunteers. Nitric Oxide 23:181–186
Lowry OH, Rosebrough NJ, Farr AL, Randall R (1951) Protein measurement with the Folin-phenol reagent. J Biol Chem 193:263–275
Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, Prabhakar NR (2011) Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J Cell Physiol 226:2925–2933
Omodeo-Sale F, Gramigna D, Campaniello R (1997) Lipid peroxidation and antioxidant systems in rat brain: effect of chronic alcohol consumption. Neurochem Res 22:577–582
Das SK, Hiran KR, Mukherjee S, Vasudevan DM (2007) Oxidative stress is the primary event: effects of ethanol consumption in brain. Indian J Clin Biochem 22:99–104
Crews FT, Nixon K (2009) Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol 44:115–127
Rump TJ, Abdul Muneer PM, Szlachetka AM, Lamb A, Haorei C, Alikunju S, Xiong H, Keblesh J, Liu J, Zimmerman MC, Jones J, Donohue TM Jr, Persidsky Y, Haorah J (2010) Acetyl-l-carnitine protects neuronal function from alcohol-induced oxidative damage in the brain. Free Radic Biol Med 49:1494–1504
Catala A (2009) Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids 157:1–11
Davis RL, Syapin PJ (2005) Interactions of alcohol and nitric-oxide synthase in the brain. Brain Res Rev 49:494–504
Olson N, van der Vliet A (2011) Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 25:125–137
Mantena SK, King AL, Andringa KK, Landar A, Darley-Usmar V, Bailey SM (2007) Novel interactions of mitochondria and reactive oxygen/nitrogen species in alcohol mediated liver disease. World J Gastroenterol 13:4967–4973
Nath B, Szabo G (2012) Hypoxia and hypoxia inducible factors: diverse roles in liver diseases. Hepatology 55:622–633
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1:393–399
Padmavathi P, Reddy VD, Maturu P, Varadacharyulu N (2010) Smoking-induced alterations in platelet membrane fluidity and Na(+)/K(+)-ATPase activity in chronic cigarette smokers. J Atheroscler Thromb 17:619–627
Wiswedel I, Gardemann A, Storch A, Peter D, Schild L (2010) Degradation of phospholipids by oxidative stress-exceptional significance of cardiolipin. Free Radic Res 44:135–145
Petrosillo G, Portincasa P, Grattagliano I, Casanova G, Matera M, Ruggiero FM, Ferri D, Paradies G (2007) Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim Biophys Acta 1767:1260–1267
Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, Schumacker PT (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 275:25130–25138
Castello PR, David PS, McClure T, Crook Z, Poyton RO (2006) Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab 3:277–287
Chong PL, Fortes PA, Jameson DM (1985) Mechanisms of inhibition of (Na, K)-ATPase by hydrostatic pressure studied with fluorescent probes. J Biol Chem 260:14484–14490
Sutherland E, Dixon BS, Leffert HL, Skally H, Zaccaro L, Simon FR (1998) Biochemical localization of hepatic surface membrane Na+, K+-ATPase activity depends on membrane lipid fluidity. Proc Nat Acad Sci 85:8673–8677
Acknowledgments
The authors thank the Director and staff of the Center for Cellular and Molecular Biology, Hyderabad, India, for providing facilities to carry out fluidity studies. This study was supported in part by the University Grants Commission (Grant No. F-3-11/97), New Delhi, India.
Conflict of interest
The authors declare that there are no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Reddy, V.D., Padmavathi, P., Kavitha, G. et al. Alcohol-induced oxidative/nitrosative stress alters brain mitochondrial membrane properties. Mol Cell Biochem 375, 39–47 (2013). https://doi.org/10.1007/s11010-012-1526-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1526-1