
Abstract
Stroke is a life-threatening disease with major cause of mortality and morbidity worldwide. The neuronal damage following cerebral ischemia is a serious risk to stroke patients. Oxidative stress and apoptotic damage play an important role in cerebral ischemic pathogenesis and may represent a target for treatment. The objective of this study was to test the hypothesis that administration of edaravone (Edv) maintains antioxidant status in brain, improves the cholinergic dysfunction and suppresses the progression of apoptosis response in rat. To test this hypothesis, male Wistar rats were subjected to middle cerebral artery occlusion (MCAO) of 2 h followed by reperfusion for 22 h. Edv was administered (10 mg/kg bwt) intraperitoneally 30 min before the onset of ischemia and 1 h after reperfusion. After reperfusion, rats were tested for neurobehavioral activities and were sacrificed for the infarct volume, estimation of oxidative damage markers. Edv treatment significantly reduced ischemic lesion volume, improved neurological deficits, contended oxidative loads, and suppressed apoptotic damage. In conclusion, treatment with Edv ameliorated the neurological and histological outcomes with elevated endogenous anti-oxidants status as well as reduced induction of apoptotic responses in MCA occluded rat. We theorized that Edv is among the pharmacological agents that reduce free radicals and its associated cholinergic dysfunction and apoptotic damage and have been found to limit the extent of brain damage following stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cerebral ischemia is a medical emergency with astronomical financial repercussions on health systems worldwide and is associated with a high incidence of sensory, motor, and cognitive deficits [1]. Ischemic stroke is caused by obstruction of blood flow to the brain, resulting in energy failure that initiates a complex series of metabolic events, ultimately causing neuronal cell death. The ensuing cascade of events causes a mitochondrial dysfunction and rapid decrease in ATP, which leads to the free radical associated cell death [2, 3].
The brain is very susceptible to the oxidative damage, due to its rapid oxidative metabolic activity, high polyunsaturated fatty acid content, relatively low anti-oxidant capacity, and inadequate neuronal cell repair activity [4, 5]. Oxidative damage to lipid, fatty acid, and protein can lead to structural and functional disruption of the cell membrane, inactivation of enzymes, and finally cell death. Earlier, our research group investigated and reported the neuroprotective effect of certain anti-oxidants on different experimental models of neurodegeneration [3, 6, 7]. Thus, treatment with anti-oxidant may boost the system to defend against the oxidative threats.
Furthermore reactive oxygen species (ROS) mediated oxidative stress is well implicated in apoptosis. Several studies have revealed that many neurons in the ischemic penumbra may undergo apoptosis after several hours or days, and thus they are potentially recoverable after the onset of stroke [8]. Apoptosis is believed to be responsible for up to 50 % of cell death during ischemia [9]. In general, there are three major mechanisms that have been proposed to account for apoptosis: mitochondrial pathways, death receptors, and endoplasmic stress. Caspases, which are implicated in all the three mechanism, plays a key role in the execution phase of apoptosis by cleaving specific proteins resulting in an irreversible commitment to cell death.
Experimental models of cerebral ischemia have been developed to improve the understanding of detrimental mechanisms involved in the ischemic brain injury, and to bone up the potential efficiency of therapeutic strategies. Among all the animal models of ischemic stroke, filamentous reversible middle cerebral artery (MCA) occlusion is one of the most widely used experimental paradigms to induce focal cerebral ischemia [3, 10, 11]. The importance of these models lies in preclinical testing of drugs designed for neuroprotection that ultimately may improve functional recovery from stroke.
Edaravone (Edv, 3-methyl-1-phenyl-2-pyrazolin-5-one) is a novel free radical scavenger with potential anti-oxidant and anti-apoptotic properties. It attenuates ischemic brain injury in patients and animal models [12, 13]. The therapeutic effects of Edv were assessed in various models of neurodegenerative diseases including stroke [13], traumatic brain injury [14], and Parkinson’s disease [15]. However, it is still unclear whether Edv plays a role in the oxidative stress associated cholinergic dysfunction following cerebral ischemia. In this study, we used a focal cerebral ischemia model to explore the effects of Edv administration on oxidative damage associated cholinergic dysfunction and apoptotic neuronal death in striatum.
Materials and methods
Chemicals and reagents
Glutathione (GSH) (oxidized and reduced), (−) epinephrine, glycine, hematoxylin, nicotinamide adenine dinucleotide phosphate reduced form (NADPH), 1-chloro-2,4-dinitrobenzene (CDNB), 5-5′-dithiobis-2-nitrobenzoic acid (DTNB), sulfosalicylic acid (SSA), thiobarbituric acid (TBA), 2,3,5-triphenyltetrazolium chloride (TTC), caspase-3 colorimetric assay kit, DAB, and Edv were purchased from Sigma-Aldrich Chemicals Private Limited, India. Bcl-2 and ChAT were purchased from BioVision, USA. Secondary anti-mouse IgG was purchased from Jackson Immuno research, USA. Other chemicals were of analytical reagent grade.
Animals and treatments
Male Wistar rats weighing 250–300 g were obtained from the Central Animal House, Jamia Hamdard, New Delhi, India. They were housed in polypropylene cages in air conditioned room and allowed free access to pellet diet and water ad libitum. The animals were used in accordance with the procedure approved by the Animal Ethics Committee of Jamia Hamdard.
Experimetal protocol
To investigate the neuroprotective effects of Edv, we used the rat middle cerebral artery occlusion (MCAO) model. Animals were divided into four groups of eight animals each. The first group served as sham and saline was given, in the second MCAO was performed, i.e., ischemia was induced for 2 h followed by reperfusion for 22 h, in the third group MCAO was performed with addition of treating the rats with Edv (i.e., Edv + MCAO group), and the fourth group was sham treated with drug alone, i.e., Edv + S group. Edv was dissolved in saline and given in a dose of 10 mg/kg intraperitoneally 30 min before the onset of ischemia. Additional injections of 10 mg/kg were administered at 1 h of post-MCAO. After completion of the reperfusion period, the animals were assessed for neurobehavioral activities and then sacrificed. The striatum was removed from the brain for biochemical estimations.
Middle cerebral artery occlusion (MCAO)
A male Wistar rat (250–300 g) overnight fasted was anaesthetized with chloral hydrate (400 mg/kg, ip) and the operative field was shaved. The right common carotid artery (CCA) was exposed and MCA was occluded using 4-0 silicone rubber coated monofilament suture as described by Khan et al. [3]. First a skin incision was made from the petrous bone to the scapula. Then, the neck muscles were dissected to gain access to the CCA. Dissection of the carotid bifurcation and glomus caroticum was performed in the vicinity of the vagus nerve. The occipital artery branching off the external carotid artery (ECA) was ligated close behind the carotid bifurcation. The internal carotid artery (ICA) was carefully dissected as distal to its entrance to the skull as possible. Proximal to this entrance, ICA gives off a branch called pterygopalatine artery, which runs more dorsal and lateral to ECA. The prepared suture was introduced through right ECA into the ICA and advanced ~17–20 mm intracranially from the CCA bifurcation to block the origin of MCA in rat. Reperfusion was done after 2 h by retraction of the nylon suture. The cut on the neck was sutured in one layer and disinfected with iodine. The animal was placed in a clean cage. In sham rats, the ECA was exposed for insertion of the filament but it was not inserted.
Infarct volume analysis
The animals were killed after 2 h of occlusion followed by 22 h of reperfusion. The brains were dissected out and kept in a brain matrix. The coronal sections of the brain of 1.5 mm was cut down in rat brain matrix with the help of sharp blades and stained with 1 % triphenyltetrazolium chloride (TTC) prepared into normal saline at 37 °C for 15 min. The infarct volume was calculated by taking the average of infarct area on both sides of the slice and multiplying it by section thickness. Infarct volumes from each section were then summed to determine total brain infarct volume and adjusted for edema.
Behavioral tests
Rotarod test
The neurological function was evaluated after reperfusion. Omni Rotor (Omnitech Electronics, Inc., Columbus, Ohio) was used to evaluate the muscular coordination skill as described by Khan et al. [3]. The animals were first trained on rota-rod before the surgery. After 22 h of the reperfusion, the animals were kept on the rotor at the speed of 10 cycles/min. The motor deficiency was evaluated as the ability of the rat to hold the rotating rotor. The time for which the rat holds the rotor was calculated and the result was expressed in seconds. Cut-off time was 180 s.
Neurological score
Neurologic deficits in the animals were assessed by an observer blinded to the identity of the groups. Neurological deficits were assessed at 24 h after reperfusion (before killing) and scored as follows: 0, no observable neurological deficit (normal); 1, failure to extend left forepaw on lifting the whole body by tail (mild); 2, circling to the contralateral side (moderate); 3, leaning to the contralateral side at rest or no spontaneous motor activity (severe). The scoring was based on the method of Masuo et al. [16].
Biochemical evaluation
Tissue preparation
After the behavioral studies, the animals were killed and their brains were removed to dissect out hippocampus and frontal cortex. The tissue at 5 % (w/v) was homogenized in 10 mM PB, pH 7.4 containing 10 μl/ml protease inhibitors [5 mM leupeptin, 1.5 mM aprotinin, 2 mM phenylethylsulfonylfluoride (PMSF), 3 mM peptastatin A, 0.1 mM EGTA, 1 mM benzamidine and 0.04 % butylated hydroxytoluene], and centrifuged at 800×g for 5 min at 4 °C. An aliquot of the supernatant (S1) was used for the assay of TBARS, while the remaining S1 was recentrifuged at 10,500×g for 15 min at 4 °C. The resulting second supernatant, the post mitochondrial supernatant (PMS), was used for the estimation of anti-oxidant enzymes and GSH.
Assay for TBARS content
The assay of TBARS was done according to method of Utley et al. [17] as described by us [18]. The homogenate 0.25 ml was incubated at 37 ± 1 °C in a metabolic shaker (120 cycles/min) for 1 h. Similarly, 0.25 ml of the same homogenate was pipetted in a test tube and incubated at 0 °C. After 1 h of incubation, 0.25 ml 5 % chilled TCA and 0.25 ml of 0.67 % TBA was added to each test tube. The mixture was centrifuged at 4,000×g for 15 min and supernatant was transferred to another tube and placed in a boiling water bath for 10 min. Thereafter, the test tubes were cooled and the absorbance of the color was read at 535 nm. The rate of lipid peroxidation was expressed as nmol TBARS formed/h/g tissue using a molar extinction coefficient of 1.56 × 105 M−1 cm−1.
Assay for reduced GSH
GSH was assayed by the method of Jollow et al. [19] with slight modification. In brief, 0.1 ml PMS was precipitated with 0.1 ml SSA (4 %). Samples were kept at 4 °C for 30 min and then subjected to centrifugation at 4,000×g for 10 min at 4 °C. The assay mixture contained 0.1 ml supernatant, 2.7 ml phosphate buffer (0.1 M, pH 7.4), and 0.2 ml DTNB (0.4 % in phosphate buffer 0.1 M, pH 7.4) in a total volume of 3.0 ml. The yellow color developed was read immediately at 412 nm using molar extinction coefficient 13.6 × 103 M−1 cm−1. The GSH content was calculated as nmol GSH/mg protein.
Assay for protein carbonyl (PC) content
PC content was measured by the method of Levine [20]. In brief, the PMS (0.5) ml was treated with an equal volume of 20 % TCA for protein precipitation. After centrifugation at 10,000×g for 10 min, the pellet was resuspended in 0.5 ml of 10 mM DNPH in 2 M HCL and kept in a dark place for 1 h by vortexing repeatedly at 10 min interval. This mixture was treated with 0.5 ml of 20 % TCA. After centrifugation at 11,000×g at 4 °C for 3 min, the precipitate was extracted three times with 0.5 ml of 10 % TCA and dissolved in 2.0 ml of NaOH at 37 °C. Absorbance was recorded at 360 nm. PC level was expressed as nmol carbonyl/mg protein, using a molar extinction of coefficient 22 × 103 M−1 cm−1.
Assay for glutathione peroxidase (GPx)
GPx activity was estimated according to the procedure described by Mohandas et al. [21]. The reaction mixture consisted of phosphate buffer (0.05 M, pH 7.0), EDTA (1 mM), sodium azide (1 mM), glutathione reductase (GR) (1 EU/ml), GSH (1 mM), NADPH (0.2 mM), hydrogen peroxide (0.25 mM), and 0.1 ml of PMS in the final volume of 2 ml. The disappearance of NADPH at 340 nm was recorded at room temperature. The enzyme activity was calculated as nmol NADPH oxidized/min/mg/protein by using molar extinction coefficient 6.22 × 103 M−1 cm−1.
Assay of Glutathione reductase (GR)
GR activity was assayed by the method of Carlberg and Mannervik [22] as modified by Mohandas et al. [21]. The assay mixture consisted of phosphate buffer (0.1 M, pH 7.6), NADPH (0.1 mM), EDTA (0.5 mM) and oxidized GSH (1 mM), and 0.05 ml of PMS in total volume of 1 ml. The enzyme activity was quantitated at room temperature by measuring the disappearance of NADPH at 340 nm and was calculated as nmol NADPH oxidized/min/mg protein using molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Assay of glutathione-S-transferase (GST)
GST activity was measured by the method of Habig et al. [23]. The reaction mixture consisted of phosphate buffer (0.1 M, pH 6.5), reduced GSH (1 mM), CDNB (1 mM), and PMS in a total volume of 1.0 ml. The change in absorbance was recorded at 340 nm and enzyme activity was calculated as nmol CDNB conjugate formed/min/mg protein using a molar extinction coefficient of 9.6 × 103 M−1 cm−1.
Glucose-6-phosphate dehydrogenase
The activity of glucose-6-phosphate dehydrogenase was assayed by the method of Zaheer et al. [24]. The reaction mixture in a total volume of 3.0 ml consisted of 0.3 ml Tris HCL buffer (0.05 M, pH 7.6), 0.1 ml NADP (0.1 mM), 0.1 ml glucose-6-phosphate (0.8 mM), 0.1 ml MgCl2 (8 mM), 0.1 ml PMS, and 2.3 ml distilled water. The change in absorbance was recorded at 340 nm and the enzyme activity was calculated as nmol NADP reduced/min/mg protein using molar extinction coefficient of 6.22 × 103 M−1 cm−1.
Western blot analysis
Western blot was performed as described previously [14]. The striatum was homogenized in lysis buffer containing 50 mM Tris–HCL pH 8.0, 50 mM NaCl, 1 % TritonX-100, and proteases cocktail. The lysate was centrifuged at 12,000×g for 10 min at 4 °C and the supernatant was collected. Equal amounts of protein were separated on 12 % SDS-polyacrylamide gels. The membrane was blocked with 5 % non-fat milk in Tris-buffered saline for 1 h. Subsequently, the membranes were incubated at 4 °C overnight with the mouse anti-ChAT antibody (1:200, Biovision). Following three washes with TBST, the membrane was incubated with the secondary horseradish peroxidase-conjugated antibody (IgG, 1:25,000) at the room temperature for 1 h. The bands were visualized by using the enhanced chemiluminescence detection reagents (Thermo scientific). Bands density was measured by densitometry and quantified using NIH-Image J software.
Immunohistochemistry
After reperfusion, the animals were anesthetized with chloral hydrate (400 mg/kg, ip) and perfused as described by Khan et al. [3] in cold phosphate buffer saline (PBS; 0.1 M, pH 7.2). The brains were removed quickly, post fixed in same fixative for 24 h, then transferred to 30 % sucrose in 0.1 M PB for at least 16 h until they sank for cryoprotection. The fixed tissues were embedded in OCT compound (polyvinyl glycol, polyvinyl alcohol, and water) and frozen at −20 °C. Coronal sections of 14 μm thickness were cut on a cryostat and collected on gelatin-coated slides and immersed in wash buffer for 20 min. Endogenous peroxidase activity was blocked with 1 % hydrogen peroxide and 10 % methanol in PBS and incubated for 30 min at room temperature. The slides were washed with PBS and the sections were overlaid with mouse anti-Bcl-2 or anti-ChAT antibodies of dilution 1:100 incubated overnight in a humid chamber at 4 °C. The slides were washed again to remove the unbound antibody and incubated with biotinylated anti-mouse IgG of dilution 1:5,000 for 1 h at room temperature. The slides were exposed to streptavidin peroxidase and the labeled sites were visualized with a solution of diaminobenzidine and hydrogen peroxide. The sections were dehydrated, cover slipped, and photomicrographs were taken.
Caspase-3 activity
The caspase-3 assay was done by colorimetric kit as given in manual (CasPASE TM colorimetric assay kit supplied with Ac-DEVD-AFC substrate).
Determination of protein
Protein was determined as described previously using bovine serum albumin (BSA) as a standard [25].
Statistical analysis
Results are expressed as mean ± SEM. Statistical analysis of the data was done by ANOVA, followed by Tukey’s Kramer test for all parameters. The p value <0.05 was considered statistically significant.
Results
Effect of Edv on infarct volume
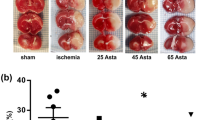
TTC staining of MCAO brain sections showed reproducible and readily detectable lesions in the areas that are supplied by the MCA after 22 h of reperfusion (Fig. 1a). The lesions were present in striatum and the overlying cortex. We hypothesized that Edv plays a protective role in stroke. Indeed, MCAO group rats have shown a significantly increased infarct volume as compared with the sham. Edv treatment has reduced the infarct volume significantly (p < 0.01) as compared with MCAO group (Fig. 1b). The infarct volume data from the present study is consistent with previously published results [39].

Edv reduces infarct volume in a rat model of MCAO. TTC-stained coronal section from each group after 2 h MCAO/22 h of reperfusion. The infarction volume was increased significantly in the MCAO group animals. Treating the animals with Edv followed by MCAO in Edv + MCAO group has decreased infarction volume as compared with MCAO group. Values are expressed as mean ± SEM (n = 8) p < 0.01, Edv + MCAO versus MCAO
Behavioral study
Rotarod was used to evaluate the muscular coordination skill of MCAO rats. Effect of MCAO on motor coordination skill and its protection by Edv is shown in Fig. 2a. Rats of MCAO group showed impairment in motor coordination skill as compared with the sham group. Treatment with Edv on MCAO group rats has significantly attenuated the motor coordination skill as compared to the MCAO group rats. No significant alteration was observed in Edv + S group rats as compared with the sham (S) group rats. Figure 2b shows a significant (p < 0.001) improvement on neurological deficit in MCAO group treated with Edv as compared with the S group animals. No significant alteration was observed in Edv + S group animals as compared with the S group animals.

Effect of Edv treatment on muscular coordination skill in MCAO rats. a MCAO leads a significant depletion in motor coordination skill as compared with the sham group and significantly recovered in Edv treated MCAO group (Edv + MCAO) as compared to MCAO group. Values are expressed as mean ± SEM for eight animals. *p < 0.001, MCAO versus sham; # p < 0.01, Edv + MCAO versus MCAO. b Average neurological scores of all rats from each group. A significant decreased neurological deficits as a functional outcome was observed in Edv + MCAO group as compared with MCAO group, indicating a profound improvement in sensory motor performance. Values are expressed as mean ± SEM of eight animals. *p < 0.001, MCAO versus sham; # p < 0.001, Edv + MCAO versus MCAO
Biochemical estimation
Effect of Edv on TBARS level
TBARS level was measured to demonstrate the oxidative damage to lipid in the striatum of MCAO group rats. Figure 3 shows the effect of Edv on TBARS level in striatum of sham and MCAO groups and its protection in Edv + MCAO group. The level of TBARS was significantly (p < 0.01) elevated in MCAO group as compared with the S group. Treating the rats with Edv significantly (p < 0.05) decreased the TBARS level in Edv + MCAO group as compared with the MCAO group. No significant change was observed in Edv + S group animals as compared with the S group animals.

Effect of Edv on TBARS level in striatum of MCAO group rats. TBARS level was significantly increased in MCAO group as compared with the sham group. Edv treatment has decreased the content of TBARS significantly in striatum of Edv + MCAO group as compared with the MCAO group. Values are expressed as mean ± SEM (n = 8). *p < 0.01 MCAO versus S, # p < 0.05 Edv + MCAO versus MCAO
Effect of Edv on GSH level
The effect of rutin on GSH content in the striatum is shown in Fig. 4. The level of GSH was depleted significantly (p < 0.01) in MCAO group animals as compared with the S group animals. Treatment with Edv in MCAO group animals has significantly (p < 0.05) protected the GSH level as compared to the MCAO group animals. No significant change was observed in Edv + S group animals as compared with the S group animals.

Effect of Edv on GSH level in striatum of MCAO group rats. GSH level was significantly decreased in MCAO group as compared with the sham group. Edv treatment has increased the content of TBARS significantly in striatum of Edv + MCAO group as compared with the MCAO group. Values are expressed as mean ± SEM (n = 8). *p < 0.01 MCAO versus S, # p < 0.05 Edv + MCAO versus MCAO
Effect of Edv on PC content
Protein oxidation was assessed by the determination of PC content in the striatum. MCAO induced a significant (p < 0.01) increase in PC content. Edv treatment significantly (p < 0.05) decreased the PC content in Edv + MCAO group. There was no statistically significant reduction in the PC content in rat treated with Edv alone in Edv + S group as compared with the S group (Fig. 5).

Effect of Edv on PC level in striatum of MCAO group rats. PC level was significantly increased in MCAO group as compared with the sham group. Edv treatment has decreased the content of PC significantly in striatum of Edv + MCAO group as compared with the MCAO group. Values are expressed as mean ± SEM (n = 8). *p < 0.01 MCAO versus S, # p < 0.05 Edv + MCAO versus MCAO
Edv attenuated the activities of anti-oxidant enzymes in striatum
The activities of anti-oxidant enzymes (GPx, GR, GST, and G6PDH) were decreased significantly in striatum of MCAO group animals as compared with the sham group animals. Edv treatment significantly increased their activities in striatum of Edv + MCAO group animals as compared with the MCAO group animals. No significant change was observed in Edv treated sham group (Edv + S) animals as compared with the S group animals (Table 1).
Effect of Edv on choline acetyltransferase (ChAT) expression
ChAT expression as measured by western blot and immunohistochemistry was analyzed to determine the cholinergic dysfunction in striatum of MCAO rats. The immunohistochemical analysis of striatum region has shown reduced expression of ChAT in MCAO group as compared with S group which was increased in Edv + MCAO group as compared with the MCAO group (Fig. 6a). Edv treatment did not show any remarkable effects in the Edv + S group compared with the S group (data not shown).

a Representative coronal brain sections of sham, MCAO, and Edv + MCAO group rats stained for ChAT. Striatum region of brain from MCAO group (B) animals show significant decreased ChAT expression as compared with the sham group (A) and this expression was increased in Edv + MCAO group (C) as compared with the MCAO group. Magnification is ×40. b Representative western blots of ChAT and β-actin from each group. Histogram shows mean densitometric analysis of bands after normalizing with β-actin as a loading control of each group. Data are mean ± SEM
We also examined the expression of ChAT by western blot. Western blot analyses showed that the MCAO brains exhibited a relatively low level of ChAT expression as compared with the sham group. However, after Edv treatment, the brain exhibited a significantly rise of ChAT expression (Fig. 6b).
Effect of Edv on Bcl2 expression
Apoptosis in ischemic stroke is mainly mediated by downregulation of Bcl2 protein expression. Figure 7 shows the protective effect of Edv on Bcl2 expression. Edv treatment increased the Bcl2 expression in striatum of Edv + MCAO group animals as compared with the MCAO group. The expression of Bcl2 was almost negligible in MCAO group as compared with the sham group. Edv treatment did not show any remarkable effects in the Edv + S group compared with the S group (data not shown).

Representative coronal brain sections of sham, MCAO, and Edv + MCAO group rats stained for Bcl-2. Striatum region of brain from MCAO group (B) animals show significant decreased Bcl-2 expression as compared with the sham group (A) and this expression was increased in Edv + MCAO group (C) as compared with the MCAO group. Magnification is ×40
Effect of Edv on caspase-3 activity
Caspase-3 is believed to be the main executioner protease of the apoptotic cascade and several models of experimental cerebral ischemia have been reported to induce the activation of caspase-3. Our result showed increased caspase-3 activity (p < 0.001) was observed in MCAO group as compared with the sham group. Caspase-3 activity was significantly (p < 0.01) attenuated by the administration of Edv in Edv + MCAO group (Fig. 8). Edv treatment did not show any remarkable effects in the Edv + S group compared with the S group.

Effect of Edv on caspase-3 activity in striatum. Caspase-3 activity was significantly increased in MCAO group as compared with the sham group. Edv treatment has decreased caspase-3 activity significantly in striatum of Edv + MCAO group as compared with the MCAO group. Values are expressed as mean ± SEM (n = 8) *p < 0.001 MCAO versus sham, # p < 0.01 Edv; MCAO versus MCAO
Discussion
The present study evaluated the neuroprotective effect of edaravone (Edv) on neurological deficit; oxidative stress associated cholinergic dysfunction and apoptotic cell death in rat model of focal cerebral ischemia. It is well documented that MCAO rodent model is an appropriate animal model used for the study of cerebral ischemia pathogenesis [3, 10, 11]. In the present study, Edv treatment significantly assuages motor deficit, biochemical, and histopathological alterations in MCAO induced rat. Neuroprotective potential of Edv suggests that it is a powerful anti-oxidant with anti-apoptotic property, corroborating previous studies [13–15, 26].
The behavioral effects are intertwined with degree of neuronal dysfunction [27]. The motor function was found to be disturbed after stroke. The behavior parameters have indicated that ischemic animals cause deterioration of motor performance along with worsen functional outcome in the rats, which are protected by treatment with Edv. Our results are in agreement with earlier studies carried out by us and other [3, 6, 28].
Infarction volume in the brain is an important determinant in assessing the consequences of ischemic stroke which leads to severe neuronal damage in the different brain parts with subsequent neurological impairment. TTC methods have been used to detect the morphological features of infarct tissue after ischemic injury [29, 30]. In the present study, MCAO group showed a prominent infarct size along with significantly altered behavioral outputs. Edv treatment not only reduced the infarct size but also improved behavioral deficits in MCAO group. Animal experiments have indicated that Edv administered after cerebral ischemia are effective in reducing infarct volume and lead to improvements in neurological outcome.
Brain has multiple sources of reactive oxygen species (ROS) [31] and a large oxidative ability, but its capacity to fight against oxidative threat is limited [36]. The occlusion followed by reperfusion caused the robust generation of free radicals which lead the lipid peroxidation, protein oxidation, and ultimately cell death. Exogenous anti-oxidant is given to scavenge the free radical to inhibit ROS and thus protect brain infarct [3, 6, 28]. MCAO leads to highly reactive ROS production increasing lipid peroxidation, which leads to cellular disintegrity and neuronal loss. Lipid peroxides and hydroperoxides cause secondary injury by further generating relatively more stable and diffusible cytotoxic agents like malondialdehyde (MDA) and 4-hydroxy-trans-2-nonenal (4-HNE), respectively, and amplify oxidative cascade [3, 32]. They react avidly with cellular nucleophiles such as GSH, and causes continuous decrease in its level through increased oxidant content or protein modification. GSH is a well-known anti-oxidant that is synthesized in the cytoplasm and is present in higher concentrations in the mitochondrial matrix. The low levels of GSH may be directly related to increased ROS, lipid peroxides, and highly reactive hydroxyl radicals [3, 6, 7].
Furthermore, GSH plays a crucial and critical role in the regulation of expression of several anti-apoptotic genes. Thus, GSH inhibition in cerebral ischemia would increase the susceptibility of plasma membranes toward peroxide attacks. However, the main cause of GSH loss during oxidative stress in brain ischemia is the formation of protein glutathione mixed disulfide (PrSSG) and loss of thiol proteins. The loss of GSH and formation of PrSSG in the brain results the various membrane dysfunction, such as inhibition on Na+ K+ ATPase activity. G6-PD maintains intracellular reduced GSH content. GSH content and activity of G6-PD has been reported to decrease in ischemic conditions [33]. We observed a significant decrease in G6-PD activity in MCAO group. Edv treatment increases the G6-PD activity.
GPx plays a predominant role in removing excess free radicals and hydroperoxides and is a major defense system against oxidative stress in the brain [34]. Evidence has been presented that the neuronal defense against H2O2, which is the most toxic molecule to the brain, is mediated primarily by the GSH system. There are several reports about the modulatory effect of Edv on lipid peroxidation, GSH, and anti-oxidant enzymes following brain injury [13, 15, 35]. In agreement with this finding, we also found that Edv significantly reduced the TBARS and PC level along with increased GSH level and activities of anti-oxidant enzymes.
ChAT is the marker enzyme for distinguishing cholinergic pathways from non-cholinergic cell groups. Cholinergic interneurons localized in the striatum are involved in motor function, cognition, and behavior. It has been well known that MCAO led to the imbalance of brain energy metabolites and excitatory amino acids. Disturbed energy metabolism is intricately associated with increased oxidative stress that disorganizes and disorients the mitochondria, resulting in a reduced supply of ATP and dysregulation of intracellular Ca2+ homeostasis and thereby limiting optimal cellular and physiological functions [41, 42]. An increase in Ca2+ influx through L-type Ca2+ channels is thought to contribute to cholinergic dysfunctions [43]. On the other hand, reduction in brain glucose below normal level also affects cholinergic system. After MCAO, striatal glucose levels immediately declined and this is likely due to consumption of local glucose and lack of supply of fresh glucose due to limited blood flow [44]. In the present study, increased ChAT protein expression level measured by western blot and immunohistochemistry may be due to oxidative stress and impaired glucose utilization in the striatum following MCAO. Edv treatment significantly restored ChAT expression by ameliorating the oxidative burdens in striatum of MCAO rat. This effect of Edv can be attributed to its potent anti-oxidant property [14, 45]. However, the exact mechanism of oxidative stress induced upregulation of ChAT protein expression is remained to be explores.
With the discovery of biochemical and histological hallmarks of apoptosis, it has become increasingly evident that the intrinsic cellular suicide program is not only required for the development of normal central nervous system, but also, if executed inappropriately, contributes to the pathology of stroke and neurodegenerative disorders [36]. Apoptotic and anti-apoptotic signaling pathways are activated after cerebral ischemia, and it is generally accepted that a shift in the balance between pro- and anti-apoptotic protein factors toward the expression of proteins that promote cell death may be one mechanism underlying apoptotic cell death [37]. Bcl-2 regulates the permeability of transition pore and thus release of cytochrome c, which is often considered as a point of no return in the subsequent caspase-3 cascade that ultimately results in apoptosis. It has been already reported that Bcl-2 expression can inhibit the generation of ROS and stabilize the mitochondrial membrane potential [14, 38]. Thus, we examined the presence of the protective Bcl-2 protein. Our study has shown with evidence that the treatment with Edv up-regulates the anti-apoptosis factors like Bcl-2 and downregulates the caspase-3 activity, thus preventing cell death in ischemic brain and leading to the recovery of neuronal tissue. This result is in agreement with other studies [39, 40].
Conclusion
In conclusion, the present study demonstrates that Edv has scavenging properties to attenuate lipid peroxidation, protein oxidation, and enhance anti-oxidant enzymes. In addition, Edv treatment limits apoptotic response and improves oxidative stress associated cholinergic dysfunction. These results suggest that the neuroprotective effect of Edv is due to the inhibition of ROS formation by scavenging free radicals and may be used for patients with acute cerebral ischemia, as well as for treating other neurodegenerative diseases.
References
Hattori K, Lee H, Hurn PD, Crain BJ, Traystman RJ, DeVries AC (2000) Cognitive deficits after focal cerebral ischemia in mice. Stroke 31:1939–1944
Shah MK, Shin W, Parikh VS, Ragin A, Mouannes J et al (2010) Quantitative cerebral MR perfusion imaging: preliminary results in stroke. J Magn Reson Imaging 32:796–802
Khan MM, Ishrat T, Ahmad A, Hoda MN, Khan MB et al (2010) Sesamin attenuates behavioral, biochemical and histological alterations induced by reversible middle cerebral artery occlusion in the rats. Chem Biol Interact 183:255–263
Halliwell B (2001) Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging 18:685–716
Cherubini A, Polidori MC, Bregnocchi M, Pezzuto S et al (2000) Antioxidant profile and early outcome in stroke patients. Stroke 31:2295–2300
Saleem S, Ahmad M, Ahmad AS, Yousuf S, Ansari MA et al (2006) Effect of saffron (Crocus sativus) on neurobehavioral and neurochemical changes in cerebral ischemia in rats. J Med Food 9:246–253
Ishrat T, Hoda MN, Khan MB, Yousuf S, Ahmad M, Khan MM et al (2009) Amelioration of cognitive deficits and neurodegeneration by curcumin in rat model of sporadic dementia of Alzheimer’s type (SDAT). Eur Neuropsychopharmacol 19:636–647
Racay P, Chomova M, Tatarkova Z, Kaplan P, Hatok J, Dobrota D (2009) Ischemia-induced mitochondrial apoptosis is significantly attenuated by ischemic preconditioning. Cell Mol Neurobiol 29:901–908
Choi DW (1996) Ischemia-induced neuronal apoptosis. Curr Opin Neurobiol 6:667–672
Takano K, Tatlisumak Y, Bergmann AG, Gibson DG, Fisher M (1997) Reproducibility and reliability of middle cerebral artery occlusion using a silicone-coated suture (Koizumi) in rats. J Neurol Sci 153:8–11
Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20:84–91
Watanabe T, Tahara M, Todo S (2008) The novel antioxidant edaravone: from bench to bedside. Cardiovasc Ther 26:101–114
Yamamoto Y, Yanagisawa M, Tak NW, Watanabe K, Takahashi C et al (2009) Repeated edaravone treatment reduces oxidative cell damage in rat brain induced by middle cerebral artery occlusion. Redox Rep 14:251–258
Wang GH, Jiang ZL, Li YC, Li X, Shi H, Gao YQ, Vosler PS, Chen J (2011) Free-radical scavenger edaravone treatment confers neuroprotection against traumatic brain injury in rats. J Neurotrauma 28:2123–2134
Yuan WJ, Yasuhara T, Shingo T, Muraoka K, Agari T et al (2008) Neuroprotective effects of edaravone-administration on 6-OHDA-treated dopaminergic neurons. BMC Neurosci 9:75
Masuo Y, Matsumoto Y, Morita S, Noguchi J (1997) A novel method for counting spontaneous motor activity in the rat. Brain Res Brain Res Protoc 1:321–326
Utley HC, Bernhein F, Hochslein P (1967) Effects of sulfhydryl reagent on peroxidation in microsomes. Arch Biochem Biophys 260:521–531
Islam F, Zia S, Sayeed I, Zafar KS, Ahmad AS (2002) Selenium induced alteration on lipids, lipid peroxidation, and thiol group in circadian rhythm centers of rat. Biol Trace Elem Res 90:1–12
Jollow DJ, Mitchell JR, Zampaghone N, Gillete JR (1974) Bromobenzene induced liver necrosis: protective role of glutathione and evidence for 3,4-bromobenzene oxide as the hepatotoxic intermediate. Pharmacology 11:161–169
Levine S (1960) Anoxic-ischemic encephalopathy in rats. Am J Pathol 36:1–17
Mohandas J, Marshall JJ, Duggin GG, Horvath JS, Tiller D (1984) Differential distribution of glutathione and glutathione related enzymes in rabbit kidneys: possible implication in analgesic neuropathy. Cancer Res 44:5086–5091
Carlberg I, Mannerviek B (1975) Glutathione reductase levels in rat brain. J Biol Chem 250:5475–5480
Habig WH, Pabst MJ, Jakoby WB (1974) Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139
Zaheer N, Tiwari KK, Krishnan PS (1965) Exposure and solubilization of hepatic mitochondrial shunt dehydrogenases. Arch Biochem Biophys 109:646–648
Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Itoh T, Satou T, Nishida S, Tsubaki M, Imano M, Hashimoto S, Ito H (2010) Edaravone protects against apoptotic neuronal cell death and improves cerebral function after traumatic brain injury in rats. Neurochem Res 35:348–355
Schwarting RK, Bonatz AE, Carey RJ, Huston JP (1991) Relationships between indices of behavioral asymmetries and neurochemical changes following mesencephalic 6-hydroxydopamine injections. Brain Res 554:46–55
Zhao J, Zhao Y, Zheng W, Lu Y, Feng G, Yu S (2008) Neuroprotective effect of curcumin on transient focal cerebral ischemia in rats. Brain Res 1229:224–232
Bederson JB, Pitts LH, Germano SM et al (1986) Evaluation of 2,3,5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke 17:1304–1308
Liszczak TM, Hedley-Whyte ET, Adams JF et al (1984) Limitation of tetrazolium salts in delineating infarcted brain. Acta Neuropathol (Berl) 65:150–157
Faraci FM (2006) Reactive oxygen species: influence on cerebral vascular tone. J Appl Physiol 100:739–743
Chang CY, Ke DS, Chen JY (2009) Essential fatty acids and human brain. Acta Neurol Taiwan 18:231–241
Sarkar S, Das N (2006) Mannosylated liposomal flavonoid in combating age-related ischemia-reperfusion induced oxidative damage in rat brain. Mech Ageing Dev 127:391–397
Imam SZ, Ali SF (2000) Selenium, an antioxidant, attenuates methamphetamine-induced dopaminergic toxicity and peroxynitrite generation. Brain Res 855:186–191
Watanabe T, Yuki S, Egawa M, Nishi H (1994) Protective effects of MCI-186 on cerebral ischemia: possible involvement of free radical scavenging and antioxidant actions. J Pharmacol Exp Ther 268:1597–1604
Cheung RT (2003) The utility of melatonin in reducing cerebral damage resulting from ischemia and reperfusion. J Pineal Res 34:153–160
Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK (2003) Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J Neurochem 85:1026–1036
Sasaki T, Kitagawa K, Yagita Y, Sugiura S, Omura-Matsuoka E et al (2006) Bcl2 enhances survival of newborn neurons in the normal and ischemic hippocampus. J Neurosci Res 84:1187–1196
Amemiya S, Kamiya T, Nito C, Inaba T, Kato K et al (2005) Anti-apoptotic and neuroprotective effects of edaravone following transient focal ischemia in rats. Eur J Pharmacol 516:125–130
Okazaki T, Magaki T, Takeda M, Kajiwara Y, Hanaya R et al (2008) Intravenous administration of bone marrow stromal cells increases survivin and Bcl-2 protein expression and improves sensorimotor function following ischemia in rats. Neurosci Lett 430:109–114
Zeevalk GD, Bernard LP, Sinha C, Ehrhart J, Nicklas WJ (1998) Excitotoxicity and oxidative stress during inhibition of energy metabolism. Dev Neurosci 20:444–453
Berridge MJ, Lipp P, Bootman MD (2000) The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol 1:11–21
Davare MA, Hell JW (2003) Increased phosphorylation of the neuronal L-type Ca(2+) channel Ca(v)1.2 during aging. Proc Natl Acad Sci U S A 100:16018–16023
Kiewert C, Mdzinarishvili A, Hartmann J, Bickel U, Klein J (2010) Metabolic and transmitter changes in core and penumbra after middle cerebral artery occlusion in mice. Brain Res 1312:101–107
Jiao L, Zhang J, Li Z, Liu H, Chen Y, Xu S (2011) Edaravone alleviates delayed neuronal death and long-dated cognitive dysfunction of hippocampus after transient focal ischemia in Wistar rat brains. Neuroscience 182:177–183
Acknowledgments
Authors are thankful to the Department of Ayurveda, Yoga and Naturalpathy, Unani, Siddha and Homeopath (AYUSH), Ministry of Health and Family Welfare, Government of India, New Delhi for financial assistance. The authors wish to thanks Mr. Dharamvir Singh for his assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ahmad, A., Moshahid Khan, M., Javed, H. et al. Edaravone ameliorates oxidative stress associated cholinergic dysfunction and limits apoptotic response following focal cerebral ischemia in rat. Mol Cell Biochem 367, 215–225 (2012). https://doi.org/10.1007/s11010-012-1335-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-012-1335-6