
Abstract
The purpose of this study was to investigate the effect of chronic treatment with etanercept (a soluble recombinant fusion protein consisting of the extracellular ligand-binding domain of tumor necrosis factor receptor type 2) on the development of hypertension in fructose-fed rats (FFR). High fructose feeding and treatment with etanercept (0.3 mg/kg, three times per week) was initiated simultaneously in male Wistar rats. Systolic blood pressure, fasted plasma parameters, insulin sensitivity, vascular reactivity, plasma angiotensin II (Ang II), and norepinephrine were determined following 9 weeks of treatment. FFR exhibited insulin resistance, hyperinsulinemia, hypertriglyceridemia, endothelial dysfunction, and hypertension. Treatment with etanercept prevented the rise in blood pressure without affecting insulin levels, insulin sensitivity, triglycerides, or Ang II levels in FFR. Etanercept treatment improved acetylcholine-induced relaxation and normalized endothelial nitric oxide synthase expression in aortas from FFR. The results of this study suggest that treatment with etanercept prevented the development of hypertension by improving vascular function and restoring endothelial nitric oxide synthase expression in FFR.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The metabolic syndrome is a clustering of cardiovascular risk factors that include abdominal obesity, dyslipidemia, insulin resistance, and hypertension. Insulin resistance has been implicated as a causal factor in the pathogenesis of the metabolic syndrome [1, 2]. Although several mechanisms have been proposed to mediate the link between insulin resistance and hypertension, insulin resistance alone does not explain the pathogenesis of this disorder. In recent years, chronic inflammation has been proposed as an important mediator in the development of insulin resistance and Type 2 diabetes [3, 4]. Numerous epidemiological studies have reported strong positive associations between systemic markers of inflammation with features of the metabolic syndrome and/or risk of cardiovascular events [4–6].
Tumor necrosis factor-α (TNF-α) is a major proinflammatory cytokine suggested to be a key mediator involved in the development of insulin resistance [7, 8]. The effects of TNF-α are mediated through its receptors: tumor necrosis factor receptor type 1 (TNFR1) or tumor necrosis factor receptor type 2 (TNFR2), with the majority of its biological actions occurring through TNFR1 activation [9]. Ligand binding to TNFR1 can result in the activation of transcription factor nuclear factor κB (NFκB) which modulates the expression of numerous inflammatory genes, including cytokines such as interleukin-6 (IL-6), chemokines, and growth factors [10, 11]. TNF-α is constitutively expressed in adipocytes and its expression is elevated in adipose tissue of obese animals [12, 13]. Studies have demonstrated that TNF-α induced insulin resistance [12–15] while inhibition of TNF-α improved insulin sensitivity in obese rodents [12, 15, 16]. In addition, elevated levels of TNF-α were associated with the development of hypertension [17–19].
Togashi et al. reported that inhibition of the TNF-α converting enzyme (TACE) improved insulin sensitivity in FFR, however, no accompanying reduction in blood pressure was observed [20]. Given that a relationship between insulin resistance and hypertension has been proposed [1, 21, 22], we found the lack of effect on blood pressure regulation surprising. This effect may be due to the experimental design utilized as treatment with a TACE inhibitor occurred only during the final 2 weeks of the 6 weeks of fructose feeding. Furthermore, TNF-α has been reported to modulate the pathways of various vasoactive mediators, such as the renin angiotensin system [23–25], however, the exact mechanism by which TNF-α modulates vasoactive mediators and contributes to hypertension secondary to insulin resistance remains to be clarified.
The aim of the present study was to investigate the effect of chronic etanercept treatment in insulin-resistant FFR. Etanercept is a soluble recombinant fusion protein consisting of the extracellular ligand-binding domain of TNFR2. Etanercept binds to and inactivates circulating TNF-α, thereby acting as a competitive inhibitor of TNF-α. We determined the effects of etanercept on plasma parameters, systolic blood pressure (SBP), insulin sensitivity, and vascular reactivity. We also examined the protein expression of downstream targets of TNF-α, such as endothelial nitric oxide synthase (eNOS), inducible nitric oxide synthase (iNOS), and NFκB. We hypothesized that treatment with etanercept will prevent the development of hypertension in FFR by improving vascular function.
Methods
Animals and experimental design
Male Wistar rats were obtained from Charles River Laboratories (St-Constant, Quebec) at 5 weeks of age and randomly divided into four experimental groups: control (C, n = 20), control etanercept-treated (CE, n = 20), fructose (F, n = 20), and fructose etanercept-treated (FE, n = 20).
At 6 weeks of age, fasted (5 h) plasma parameters (glucose, insulin and triglycerides) and SBP were measured in all groups. At 7 weeks of age, rats in fructose-fed groups (F and FE) were started on a 60% fructose diet (Teklad Laboratory Diets, Madison, WI) for 6 weeks, whereas rats in control groups (C and CE) were maintained on standard laboratory rat chow containing 30% carbohydrate in the form of starch for the same period. Treatment with etanercept (CE and FE) was initiated concurrently at a dose of 0.3 mg/kg/day, three times per week via s.c. injection for the duration of the study. The dose of etanercept was used based on effective inhibition of TNF-α from previous studies in rats [26–28]. Rats were housed on a 12 h light–dark cycle and received food and water ad libitum. At the end of the study, rats were euthanized with an overdose of pentobarbital (65 mg/kg, i.p.). The thoracic aorta, first branch order of the superior mesenteric artery and epidydimal fat pads were isolated, cleaned of adherent connective tissue, and used in further studies.
This investigation conforms with the Canadian Council on Animal Care Guidelines on the Care and Use of Experimental Animals. All protocols were approved by the University of British Columbia Animal Care Committee.
Blood pressure measurements
Prior to obtaining blood pressure measurements, rats were preconditioned to the procedure. SBP was measured in conscious rats using the indirect non-invasive tail-cuff method without external preheating as previously described [29, 30].
Oral glucose tolerance test and insulin sensitivity index
After 9 weeks of study, rats were fasted overnight (15 h) and subjected to an oral glucose tolerance test (OGTT). A 40% glucose solution was prepared and administered by oral gavage (1 g/kg) to conscious animals. Blood samples were obtained at 0, 10, 20, 30, 60, and 90 min following the glucose challenge. Plasma was separated and stored at −20°C until further analysis. The insulin sensitivity index was calculated for each animal using data obtained from the OGTT using the formula of Matsuda and DeFronzo where insulin sensitivity index = 100/square root [(mean plasma glucose × mean plasma insulin) × (fasting plasma glucose × fasting plasma insulin)] [31]. Values obtained through an OGTT correlate highly with results obtained from the euglycemic hyperinsulinemic clamp technique [31].
Blood collection
Five hour-fasted blood samples were collected from the tail vein for the determination of plasma glucose, insulin, triglycerides, TNF-α, and IL-6 levels. Plasma was separated, aliquoted, and stored at−20°C until further analysis. At termination, blood was collected via cardiac puncture for assaying angiotensin II (Ang II) or norepinephrine (NE). For plasma Ang II determination, blood was aliquoted into plastic tubes containing 0.44 mM o-phenanthroline, 25 mM ethylenediaminetetraacetic acid, 1 mM p-hydroxymercuribenzoic acid, and 0.12 mM pepstatin A. For determination of plasma NE levels, blood was aliquoted into heparinized tubes. Plasma was separated and stored at−80°C until analysis.
Vascular reactivity
Isolated superior mesenteric arteries were cut into 3 mm rings and suspended on wire hooks in isolated tissue baths containing modified Krebs-Ringer bicarbonate solution pH 7.4 (118 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 0.026 mM edentate calcium disodium (EDTA), 11.1 mM glucose, and 25 mM NaHCO3) aerated with 95% O2: 5% CO2 at 37°C as previously described [32, 33]. Resting tension was adjusted to 1.0 g for each ring to allow for maximal active force generated. Changes in vascular tension were recorded using a force transducer on a Grass polygraph machine (model 79D). After an equilibration period, rings were challenged with 40 mM KCl and endothelial integrity was assessed. Vessels were subjected to a cumulative dose–response curve to phenylephrine (PE; 10−9 to 10−4 mol/l) followed by a cumulative dose–response curve to acetylcholine (ACh; 10−9 to 10−4 mol/l) in arteries precontracted with the ED70 of PE. Vessels were equilibrated to baseline between dose–response curves.
PE-induced responses were expressed as a percentage of the maximal response to KCl. Changes in ACh-induced responses were expressed as a percentage of the response to PE in each tissue. The maximum response (E max) and negative log of the concentration producing 50% of the maximum response (pD 2) were obtained by nonlinear regression analysis of individual dose–response curves using GraphPad Prism, version 5.0 (GraphPad Software, Inc., San Diego, CA).
Western blot analysis
Thoracic aorta and epidydimal fat pads were frozen in liquid nitrogen, powdered, and homogenized in RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM PMSF, 1 mM EDTA, 10 μg/ml aprotinin, 5 μg/ml leupeptin, 2.5 μg/ml pepstatin, 0.5% sodium deoxycholate, 0.1% SDS). Homogenates were centrifuged at 12 000g for 15 min and supernatants were collected. Total protein content was determined using the Bradford protein assay. Equal amounts of protein were subjected to sodium dodecyl polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were transferred onto polyvinylidene difluoride membrane. Membranes were blocked with 5% non-fat milk in Tris-buffered saline-Tween (TBS-T) and incubated with the appropriate primary antibody. Immune complexes were detected using horseradish peroxidase conjugated secondary antibody for 1 h at room temperature (5% milk/TBS-T) and a chemiluminescence detection kit (Amersham Pharmacia). Band intensity was analyzed using densitometry and represented as a percent of the control mean.
Biochemical measurements
Plasma glucose levels were determined using a Beckman Glucose Analyzer II (Beckman, Fullerton, CA). Plasma triglycerides were measured using an enzymatic colourimetric assay from Boehringer Mannheim (Germany). Plasma insulin levels were determined using a radioimmunoassay kit from Linco Research (St. Charles, MO). Plasma Ang II and NE levels were measured using an enzyme immunoassay kit from Cedarlane (Hornby, Ontario) and IBL Hamburg (Toronto, Ontario), respectively. Plasma TNF-α and IL-6 were determined using enzyme immunoassay kits from Biosource (Camarillo, CA).
Reagents
All chemicals were reagent grade and purchased from Sigma (St. Louis, MO). Solutions of PE and ACh were freshly prepared in Krebs-Ringer buffer. Etanercept was a generous gift from Amgen (Seattle, WA).
Statistical analysis
All data are expressed as mean ± SEM. Statistical analysis of all data was performed using the Number Cruncher Statistical Software 2000 (NCSS, Kaysville, UT). Data with multiple time points were analyzed by General Linear Model ANOVA and inter-group comparisons of dependent variables were analyzed by one-way ANOVA. For all results, the Newman-Keuls test for post-hoc analysis was applied. A value of P < 0.05 was taken as the level of significance.
Results
General characteristics, SBP, and insulin sensitivity index
General characteristics of rats following 9 weeks of etanercept treatment are summarized in Table 1. No differences in body weight or fasted plasma glucose were observed among the experimental groups. Food intake was slightly, but significantly, reduced in fructose-fed (F and FE) animals as compared to control (C and CE) animals. Fasted plasma insulin and triglyceride levels were significantly elevated in FFR. Treatment with etanercept had no effect on any parameters measured in either control or FFR. Following 9 weeks of study, animals fed a high fructose diet had significantly elevated SBP. Chronic treatment with etanercept prevented the increase in blood pressure in FFR. Treatment with etanercept had no effect on blood pressure in control rats. High fructose feeding resulted in significantly impaired insulin sensitivity as demonstrated by the decreased insulin sensitivity index values (Table 1). Chronic etanercept treatment did not alter insulin sensitivity in either control or FFR.
Vascular reactivity
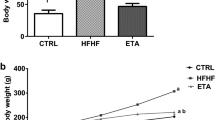
PE-induced contractile responses of superior mesenteric arteries from control and FFR are shown in Fig. 1a. There were no differences in the maximum contractile response (E max) or sensitivity (pD 2) to PE (Table 2). Chronic etanercept treatment had no effect on vessels from either control or FFR. ACh-induced relaxation of mesenteric arteries from animals following 9 weeks of treatment are shown in Fig. 1b. Arteries from FFR had significantly impaired relaxation responses to ACh as compared to arteries from control rats. Etanercept treatment normalized E max values of arteries from FFR without altering the sensitivity to ACh (Table 2). Etanercept treatment had no effect on vessels from control animals.

Cumulative dose-response curves to a PE and b ACh in superior mesenteric arteries from control and FFR rats following 9 weeks of etanercept treatment. Values expressed as mean ± SEM, n = 14
Plasma TNF-α and IL-6 levels
Plasma TNF-α were significantly elevated in control and FFR treated with etanercept following 9 weeks of treatment (Fig. 2a). There was a trend for animals from the F group to have slightly elevated levels of plasma TNF-α as compared to animals in the C group, although this increase did not reach statistical significance. No difference was observed in plasma levels of IL-6 after 9 weeks of treatment (Fig. 2b).

Effect of chronic etanercept treatment on plasma a TNF-α (n = 20) and b IL-6 (n = 12) levels in control and FFR following 9 weeks of study. Values expressed as mean ± SEM, n = 20. ‡ P < 0.05 vs. C; * P < 0.05 vs. F
Plasma NE and Ang II levels
Plasma NE levels were statistically significant among the experimental groups, however, the Newman-Keuls post-hoc test was unable to determine where the differences were (Fig. 3a). Chronic etanercept treatment had a tendency to increase NE levels in both control and FFR. Plasma Ang II levels were significantly elevated following 9 weeks of high fructose feeding (Fig. 3b). Chronic etanercept treatment had no effect on plasma Ang II in either control or FFR.

Effect of chronic etanercept treatment on plasma a NE (n = 12) and b Ang II (n = 20) levels in control and FFR following 9 weeks of study. Values expressed as mean ± SEM, n = 12. † P < 0.05 vs. C, CE
Western blot analysis
Arteries from FFR had significantly reduced protein expression of eNOS as compared to arteries from rats in the C and CE groups (Fig. 4b). Animals from the FE group showed significantly increased eNOS expression as compared to arteries from the F group. No significant differences were observed in the expression of iNOS among the experimental groups (Fig. 4d).

Representative Western blots of a eNOS and c iNOS from thoracic aorta from control and FFR following 9 weeks of study. Effect of chronic etanercept treatment on the relative intensity of b eNOS and d iNOS protein expression in control and FFR following 9 weeks of study. Values expressed as mean ± SEM, n = 10. † P < 0.05 different from C, CE; *P < 0.05 vs. F
There was a trend for fat pads from FFR to have increased protein expression of transmembrane TNF-α, although this difference did not reach statistical significance (Fig. 5b). Fat pads from animals in the FE group exhibited significant elevations in transmembrane TNF-α as compared to animals from the C and CE groups. Although there was a trend for fat pads from the F group to have increased protein expression of soluble TNF-α (Fig. 4d), this difference did not reach statistical significance.

Representative Western blots of a transmembrane TNF-α (26 kDa) and c soluble TNF-α (17 kDa) from epidydimal fat pads from control and FFR following 9 weeks of study. Effect of chronic etanercept treatment on the relative intensity of b transmembrane TNF-α (26 kDa), and d soluble TNF-α (17 kDa) protein expression in control and FFR following 9 weeks of study. Values expressed as mean ± SEM, n = 12. * P < 0.05 vs. C, CE
Fat pads from animals in the CE, F, and FE groups had significantly reduced phosphorylation of the p65 subunit of NFκB, indicating a decreased activation of NFκB p65, as compared to animals in the C group (Fig. 6b). There were no significant differences observed in the expression of the p100 subunit of NFκB which is the precursor to the p52 subunit of NFκB (Fig. 6d). No differences were observed in the expression of the p52 subunit of NFκB, which is a necessary subunit to NFκB, among the experimental groups (Fig. 6f).

Representative Western blots of a p-NFκB, c NFκB p100, and e NFκB p52 from epidydimal fat pads from control and FFR following 9 weeks of study. Effect of chronic etanercept treatment on the relative intensity of b p-NFκB, d NFκB p100, and f NFκB p52 protein expression in control and FFR following 9 weeks of study. Values expressed as mean ± SEM, n = 12. ‡ P < 0.05 vs. C
Discussion
High fructose feeding resulted in insulin resistance, hyperinsulinemia, hypertriglyceridemia, and elevated SBP. Chronic treatment with etanercept, a soluble recombinant fusion protein consisting of the extracellular ligand binding domain of TNFR2, prevented the rise in SBP, while normalizing eNOS expression and vascular function. Surprisingly, it did so without affecting insulin levels, insulin sensitivity, or triglyceride levels in these animals. Given the lack of elevated plasma TNF-α levels in FFR, and that etanercept treatment was not associated with an improvement in insulin sensitivity in these animals, its ability to prevent the rise in blood pressure was unexpected. We propose two potential mechanisms that may explain these findings. It is possible an elevation in TNF-α occurred as an initiating event that subsequently activated other vasoactive mediators, contributing to the eventual development of hypertension. Alternatively, etanercept may be acting through a mechanism independent of TNF-α inhibition.
An association between TNF-α and insulin resistance has been demonstrated in animal models of obesity and insulin resistance [12, 15, 16]. Togashi et al. demonstrated that FFR had elevated TNF-α levels and treatment with a TACE inhibitor restored insulin sensitivity [20]. In contrast, we found that following 9 weeks of fructose feeding, FFR did not exhibit elevations in inflammatory mediators, TNF-α, IL-6, or iNOS, which suggests that the development of fructose-induced hypertension is not associated with a maintained inflammatory-mediated process. Interestingly, there was a significant decrease in the phosphorylation of NFκB p65 in FFR as compared to control animals, which suggests that inflammation is actually reduced in FFR. A possible explanation for these observations is that an elevation in TNF-α levels occurs early after the onset of fructose feeding, and that subsequent compensatory mechanisms are activated to negatively regulate secretion of TNF-α into the circulation. The concept of reverse signalling has recently emerged and proposes that binding of transmembrane TNF-α to its receptor can transduce a reverse signal through the cytoplasmic tail of transmembrane TNF-α triggering cell activation, cytokine suppression, or apoptosis of the cell containing the transmembrane TNF-α [34]. However, if early elevations in TNF-α were occurring in FFR and initiating a sequence of events leading to the eventual development of insulin resistance, it is not clear why treatment with etanercept did not prevent a decrease in insulin sensitivity in these animals. It is possible that in this non-obese model, TNF-α is not the major factor in the development of insulin resistance. Alternatively, etanercept may not be able to prevent the effects of TNF-α on the insulin signalling pathway. It should be noted that in contrast to insulin-resistant, obese rodents [12, 15, 16], etanercept treatment in humans resulted in no improvements in insulin sensitivity in patients with the metabolic syndrome [35, 36], insulin resistance [37, 38] or Type 2 diabetes [39]. It is possible that in humans, etanercept is not capable of altering the site at which TNF-α interrupts the insulin signalling pathway [39], or as a result of the autocrine/paracrine action of TNF-α, neutralizing circulating TNF-α may be insufficient to improve insulin action [38]. Alternatively, the dissociation of soluble TNF-α from etanercept may allow soluble TNF-α to remain biologically active and able to signal through its cell surface receptors [40].
As expected, etanercept treatment reduced NFκB activation and increased plasma TNF-α in both control and FFR. Similar increases in TNF-α have been observed in humans treated with etanercept [41–43] and may be attributed to the ability of etanercept to prolong the half-life of TNF-α by acting as a tumour necrosis factor carrier [44, 45]. Eason et al. have demonstrated that elevated levels of TNF-α during etanercept treatment were associated with concomitant low levels of TNF-α bioactivity [46], which may explain why we observed increased TNF-α levels paralleled with decreased activation of NFκB.
To the best of our knowledge, this is the first study to demonstrate that etanercept treatment prevented the development of hypertension, normalized acetylcholine-induced relaxation, and restored eNOS expression in FFR. Our findings are consistent with previous studies that have reported improved acetylcholine-induced relaxation with etanercept treatment [27, 28, 47–49] and improved eNOS expression following treatment with a neutralizing antibody to TNF-α [50]. However, in these studies, the beneficial effects of etanercept on endothelial function and eNOS expression have been associated with its ability to inhibit TNF-α. More recently, the role of increased oxidative stress as a proposed mechanism in the development of fructose-induced hypertension has gained attention [51, 52]. Interestingly, in a rodent model of Ang II-induced hypertension, vascular infiltration of T lymphocytes associated with an increase in reactive oxygen species production was demonstrated, while treatment with etanercept not only prevented the increase in vascular oxidative stress, it also prevented the development of hypertension [53]. In the present study, we found that Ang II levels were elevated in both untreated and etanercept-treated FFR. We propose that the beneficial effects of etanercept on blood pressure, vascular reactivity, and eNOS protein expression may have occurred through a reduction in oxidative stress. Whether this is due to inhibition of the effects of locally produced TNF-α, or to a TNF-α-independent mechanism is not clear. However, the ability of etanercept to reduce blood pressure and improve vascular function despite its lack of effect on insulin resistance may be of clinical importance in conditions such as the metabolic syndrome or Type 2 diabetes and requires further investigation.
Hypertriglyceridemia, and not insulin resistance/hyperinsulinemia, has been proposed as a causal link in the development of hypertension in FFR [54]. Our data do not support this hypothesis given that the development of hypertension was attenuated in FFR chronically treated with etanercept, despite the presence of hypertriglyceridemia. Similar observations have been made in FFR treated with the AT1 receptor antagonists, losartan [55], TCV-116 [56] or L-158,809 [57], or thromboxane synthase inhibitor, dazmegral [58] in which the development of hypertension was prevented despite the presence of hypertriglyceridemia. Furthermore, a reduction in elevated triglyceride levels did not reduce elevated blood pressure in FFR [59, 60], supporting the concept of a triglyceride-independent component involved in the development of fructose-induced hypertension.
In conclusion, results from the present study suggest that chronic etanercept treatment prevented the development of hypertension by improving vascular function and restoring eNOS expression in FFR. These effects do not appear to be associated with a maintained state of chronic inflammation and occurred in an insulin-independent manner.
References
Reaven GM (1988) Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 37:1595–1607. doi:10.2337/diabetes.37.12.1595
Ginsberg HN (2000) Insulin resistance and cardiovascular disease. J Clin Invest 106:453–458. doi:10.1172/JCI10762
Pickup JC, Crook MA (1998) Is type II diabetes mellitus a disease of the innate immune system? Diabetologia 41:1241–1248. doi:10.1007/s001250051058
Festa A, D’Agostino R Jr, Howard G, Mykkanen L, Tracy RP, Haffner SM (2000) Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 102:42–47
Pickup JC, Mattock MB, Chusney GD, Burt D (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40:1286–1292. doi:10.1007/s001250050822
Frohlich M, Imhof A, Berg G, Hutchinson WL, Pepys MB, Boeing H, Muche R, Brenner H, Koenig W (2000) Association between C-reactive protein and features of the metabolic syndrome: a population-based study. Diabetes Care 23:1835–1839. doi:10.2337/diacare.23.12.1835
Moller DE (2000) Potential role of TNF-alpha in the pathogenesis of insulin resistance and type 2 diabetes. Trends Endocrinol Metab 11:212–217. doi:10.1016/S1043-2760(00)00272-1
Borst SE (2004) The role of TNF-alpha in insulin resistance. Endocrine 23:177–182. doi:10.1385/ENDO:23:2-3:177
Tracey D, Klareskog L, Sasso EH, Salfeld JG, Tak PP (2008) Tumor necrosis factor antagonist mechanisms of action: a comprehensive review. Pharmacol Ther 117:244–279. doi:10.1016/j.pharmthera.2007.10.001
Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18:2195–2224. doi:10.1101/gad.1228704
Perkins ND (2007) Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol 8:49–62. doi:10.1038/nrm2083
Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259:87–91. doi:10.1126/science.7678183
Hofmann C, Lorenz K, Braithwaite SS, Colca JR, Palazuk BJ, Hotamisligil GS, Spiegelman BM (1994) Altered gene expression for tumor necrosis factor-alpha and its receptors during drug and dietary modulation of insulin resistance. Endocrinology 134:264–270. doi:10.1210/en.134.1.264
Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM (1994) Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA 91:4854–4858. doi:10.1073/pnas.91.11.4854
Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS (1997) Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature 389:610–614. doi:10.1038/39335
Cheung AT, Ree D, Kolls JK, Fuselier J, Coy DH, Bryer-Ash M (1998) An in vivo model for elucidation of the mechanism of tumor necrosis factor-alpha (TNF-alpha)-induced insulin resistance: evidence for differential regulation of insulin signaling by TNF-alpha. Endocrinology 139:4928–4935. doi:10.1210/en.139.12.4928
Zinman B, Hanley AJ, Harris SB, Kwan J, Fantus IG (1999) Circulating tumor necrosis factor-alpha concentrations in a native Canadian population with high rates of type 2 diabetes mellitus. J Clin Endocrinol Metab 84:272–278. doi:10.1210/jc.84.1.272
Ito H, Ohshima A, Tsuzuki M, Ohto N, Takao K, Hijii C, Yanagawa M, Ogasawara M, Nishioka K (2001) Association of serum tumour necrosis factor-alpha with serum low-density lipoprotein-cholesterol and blood pressure in apparently healthy Japanese women. Clin Exp Pharmacol Physiol 28:188–192. doi:10.1046/j.1440-1681.2001.03429.x
Bautista LE, Vera LM, Arenas IA, Gamarra G (2005) Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens 19:149–154. doi:10.1038/sj.jhh.1001785
Togashi N, Ura N, Higashiura K, Murakami H, Shimamoto K (2002) Effect of TNF-alpha-converting enzyme inhibitor on insulin resistance in fructose-fed rats. Hypertension 39:578–580. doi:10.1161/hy0202.103290
DeFronzo RA (1992) Insulin resistance, hyperinsulinemia, and coronary artery disease: a complex metabolic web. J Cardiovasc Pharmacol 20(Suppl 11):S1–S16. doi:10.1097/00005344-199200111-00002
Bhanot S, McNeill JH (1996) Insulin and hypertension: a causal relationship? Cardiovasc Res 31:212–221
Antonipillai I, Wang Y, Horton R (1990) Tumor necrosis factor and interleukin-1 may regulate renin secretion. Endocrinology 126:273–278
Brasier AR, Li J, Wimbish KA (1996) Tumor necrosis factor activates angiotensinogen gene expression by the Rel A transactivator. Hypertension 27:1009–1017
Gurantz D, Cowling RT, Villarreal FJ, Greenberg BH (1999) Tumor necrosis factor-alpha upregulates angiotensin II type 1 receptors on cardiac fibroblasts. Circ Res 85:272–279
Joussen AM, Poulaki V, Mitsiades N, Kirchhof B, Koizumi K, Dohmen S, Adamis AP (2002) Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. FASEB J 16:438–440
Arenas IA, Armstrong SJ, Xu Y, Davidge ST (2005) Chronic tumor necrosis factor-alpha inhibition enhances NO modulation of vascular function in estrogen-deficient rats. Hypertension 46:76–81. doi:10.1161/01.HYP.0000168925.98963.ef
Arenas IA, Armstrong SJ, Xu Y, Davidge ST (2006) Tumor necrosis factor-alpha and vascular angiotensin II in estrogen-deficient rats. Hypertension 48:497–503. doi:10.1161/01.HYP.0000235865.03528.f1
Bunag RD (1973) Validation in awake rats of a tail-cuff method for measuring systolic pressure. J Appl Physiol 34:279–282
Hwang IS, Ho H, Hoffman BB, Reaven GM (1987) Fructose-induced insulin resistance and hypertension in rats. Hypertension 10:512–516
Matsuda M, DeFronzo RA (1999) Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care 22:1462–1470. doi:10.2337/diacare.22.9.1462
Verma S, Bhanot S, Yao L, McNeill JH (1996) Defective endothelium-dependent relaxation in fructose-hypertensive rats. Am J Hypertens 9:370–376. doi:10.1016/0895-7061(95)00392-4
Verma S, Bhanot S, McNeill JH (1996) Decreased vascular reactivity in metformin-treated fructose-hypertensive rats. Metabolism 45:1053–1055. doi:10.1016/S0026-0495(96)90000-1
Eissner G, Kirchner S, Lindner H, Kolch W, Janosch P, Grell M, Scheurich P, Andreesen R, Holler E (2000) Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J Immunol 164:6193–6198
Bernstein LE, Berry J, Kim S, Canavan B, Grinspoon SK (2006) Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med 166:902–908. doi:10.1001/archinte.166.8.902
Lo J, Bernstein LE, Canavan B, Torriani M, Jackson MB, Ahima RS, Grinspoon SK (2007) Effects of TNF-alpha neutralization on adipocytokines and skeletal muscle adiposity in the metabolic syndrome. Am J Physiol Endocrinol Metab 293:E102–E109. doi:10.1152/ajpendo.00089.2007
Ofei F, Hurel S, Newkirk J, Sopwith M, Taylor R (1996) Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45:881–885. doi:10.2337/diabetes.45.7.881
Paquot N, Castillo MJ, Lefebvre PJ, Scheen AJ (2000) No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85:1316–1319. doi:10.1210/jc.85.3.1316
Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A, Torp-Pedersen C (2005) Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res 42:517–525. doi:10.1159/000088261
Scallon B, Cai A, Solowski N, Rosenberg A, Song XY, Shealy D, Wagner C (2002) Binding and functional comparisons of two types of tumor necrosis factor antagonists. J Pharmacol Exp Ther 301:418–426. doi:10.1124/jpet.301.2.418
Tsimberidou AM, Waddelow T, Kantarjian HM, Albitar M, Giles FJ (2003) Pilot study of recombinant human soluble tumor necrosis factor (TNF) receptor (p75) fusion protein (TNFR:Fc; Enbrel) in patients with refractory multiple myeloma: increase in plasma TNF alpha levels during treatment. Leuk Res 27:375–380. doi:10.1016/S0145-2126(02)00082-6
Madhusudan S, Foster M, Muthuramalingam SR, Braybrooke JP, Wilner S, Kaur K, Han C, Hoare S, Balkwill F, Talbot DC, Ganesan TS, Harris AL (2004) A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin Cancer Res 10:6528–6534. doi:10.1158/1078-0432.CCR-04-0730
Nowlan ML, Drewe E, Bulsara H, Esposito N, Robins RA, Tighe PJ, Powell RJ, Todd I (2006) Systemic cytokine levels and the effects of etanercept in TNF receptor-associated periodic syndrome (TRAPS) involving a C33Y mutation in TNFRSF1A. Rheumatology (Oxford) 45:31–37. doi:10.1093/rheumatology/kei090
Mohler KM, Torrance DS, Smith CA, Goodwin RG, Stremler KE, Fung VP, Madani H, Widmer MB (1993) Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol 151:1548–1561
Evans TJ, Moyes D, Carpenter A, Martin R, Loetscher H, Lesslauer W, Cohen J (1994) Protective effect of 55- but not 75-kD soluble tumor necrosis factor receptor-immunoglobulin G fusion proteins in an animal model of gram-negative sepsis. J Exp Med 180:2173–2179. doi:10.1084/jem.180.6.2173
Eason JD, Pascual M, Wee S, Farrell M, Phelan J, Boskovic S, Blosch C, Mohler KM, Cosimi AB (1996) Evaluation of recombinant human soluble dimeric tumor necrosis factor receptor for prevention of OKT3-associated acute clinical syndrome. Transplantation 61:224–228. doi:10.1097/00007890-199601270-00011
Arenas IA, Xu Y, Davidge ST (2006) Age-associated impairment in vasorelaxation to fluid shear stress in the female vasculature is improved by TNF-alpha antagonism. Am J Physiol Heart Circ Physiol 290:H1259–H1263. doi:10.1152/ajpheart.00990.2005
Csiszar A, Labinskyy N, Smith K, Rivera A, Orosz Z, Ungvari Z (2007) Vasculoprotective effects of anti-tumor necrosis factor-alpha treatment in aging. Am J Pathol 170:388–398. doi:10.2353/ajpath.2007.060708
Fichtlscherer S, Rossig L, Breuer S, Vasa M, Dimmeler S, Zeiher AM (2001) Tumor necrosis factor antagonism with etanercept improves systemic endothelial vasoreactivity in patients with advanced heart failure. Circulation 104:3023–3025. doi:10.1161/hc5001.101749
Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C (2006) Tumor necrosis factor-alpha induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res 99:69–77. doi:10.1161/01.RES.0000229685.37402.80
Delbosc S, Paizanis E, Magous R, Araiz C, Dimo T, Cristol JP, Cros G, Azay J (2005) Involvement of oxidative stress and NADPH oxidase activation in the development of cardiovascular complications in a model of insulin resistance, the fructose-fed rat. Atherosclerosis 179:43–49. doi:10.1016/j.atherosclerosis.2004.10.018
Song D, Hutchings S, Pang CC (2005) Chronic N-acetylcysteine prevents fructose-induced insulin resistance and hypertension in rats. Eur J Pharmacol 508:205–210. doi:10.1016/j.ejphar.2004.12.018
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG (2007) Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204:2449–2460. doi:10.1084/jem.20070657
Si X, Webb RC, Richey JM (1999) Bezafibrate, an anti-hypertriglyceridemic drug, attenuates vascular hyperresponsiveness and elevated blood pressure in fructose-induced hypertensive rats. Can J Physiol Pharmacol 77:755–762. doi:10.1139/cjpp-77-10-755
Navarro-Cid J, Maeso R, Perez-Vizcaino F, Cachofeiro V, Ruilope LM, Tamargo J, Lahera V (1995) Effects of losartan on blood pressure, metabolic alterations, and vascular reactivity in the fructose-induced hypertensive rat. Hypertension 26:1074–1078
Chen S, Noguchi Y, Izumida T, Tatebe J, Katayama S (1996) A comparison of the hypotensive and hypoglycaemic actions of an angiotensin converting enzyme inhibitor, an AT1a antagonist and troglitazone. J Hypertens 14:1325–1330. doi:10.1097/00004872-199611000-00011
Tran LT, Macleod KM, McNeill JH (2009) Endothelin-1 modulates angiotensin II in the development of hypertension in fructose-fed rats. Mol Cell Biochem 325:89–97
Galipeau D, Arikawa E, Sekirov I, McNeill JH (2001) Chronic thromboxane synthase inhibition prevents fructose-induced hypertension. Hypertension 38:872–876
Galipeau D, Verma S, McNeill JH (2002) Female rats are protected against fructose-induced changes in metabolism and blood pressure. Am J Physiol Heart Circ Physiol 283:H2478–H2484
Fujioka Y, Masai M, Tsuboi S, Okumura T, Morimoto S, Tsujino T, Ohyanagi M, Iwasaki T (2003) Troglitazone reduces activity of the Na+/H+ exchanger in fructose-fed borderline hypertensive rats. Hypertens Res 26:111–116. doi:10.1291/hypres.26.111
Acknowledgments
This work was supported by the Heart and Stroke Foundation of British Columbia and Yukon. LTT was the recipient of a Graduate Research Scholarship in Pharmacy from the Health Research Foundation of Canada’s Research-Based Pharmaceutical Companies and the Canadian Institute for Health Research and a Pacific Century Graduate Scholarship from the University of British Columbia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tran, L.T., MacLeod, K.M. & McNeill, J.H. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem 330, 219–228 (2009). https://doi.org/10.1007/s11010-009-0136-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-009-0136-z