
Abstract
Although subclinical inflammation and oxidative stress are implicated in the aetiology of diabetes, there are hardly any studies in prediabetes. Therefore, we made an attempt to study the gene expression pattern of certain inflammatory/oxidative genes using lymphocytes from Type 2 diabetic patients, impaired glucose tolerance (IGT), and normal glucose tolerance (NGT) subjects. Compared to NGT group, interleukin-6, tumor necrosis factor-α (TNF-α), p22Phox NADPH oxidase, and thioredoxin interacting protein (TXNIP) mRNA levels were higher and suppressor of cytokine signaling (SOCS-3) mRNA was lower in subjects with IGT and diabetes. The mean (±SE) levels of thiobarbituric acid reactive substances and protein carbonyl content were also elevated in glucose intolerant subjects. In multiple linear regression analysis, TXNIP and TNF-α showed a significant association with HbA1c even after adjusting for TBARS and PCO (TXNIP: β = 1.70, P < 0.01; TNF-α: β = 1.86, P < 0.01). Increased subclinical inflammation/oxidation is seen in Asian Indians with not only Type 2 diabetes but also IGT.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Type 2 diabetes is a global health crisis of epidemic proportions that exacts an enormous toll on public health. Diabetes has detrimental effects on virtually all physiological systems. As its underlying molecular mechanisms are elucidated, it has become clear that hyperglycemia has profound effects on immunity, inflammatory function, and redox balance. Convincing data in the literature demonstrate alterations in inflammation and redox balance in Type 2 diabetes, with elevation of systemic markers in diabetic subjects, including interleukin (IL-6), tumour necrosis factor (TNF-α), soluble TNF-αR, C-reactive protein, and various oxidative stress fingerprints [1–4].
Since a large percentage of individuals with Type 2 diabetes pass through a period of prediabetes [5], it is important to know whether subclinical inflammation or oxidation associates with impaired glucose tolerance (IGT). In fact, this has been suggested in many studies that looked at systemic markers of inflammation and oxidative stress [6, 7]. Genomic analysis of skeletal muscle samples from patients with diabetes mellitus has revealed the reduced expression of genes encoding key enzymes in oxidative metabolism and mitochondrial function [8]. Moreover, the same pattern of gene expression is also observed in prediabetic individuals. Despite the fact that prediabetes is a continuum of pathological consequences in the natural history of Type 2 diabetes, there are very few studies that looked at gene expression patterns in individuals with IGT.
We have chosen to study peripheral blood lymphocytes with the hypothesis that alterations in inflammatory function and redox balance associated with Type 2 diabetes would be manifested in the peripheral immune system. Given relatively easy access to peripheral blood cells compared to target tissues of insulin action, study of peripheral immune cells would expedite research into mechanisms of immune dysfunction and oxidative reactions in Type 2 diabetes [9–12]. We studied lymphocytes because they equally contribute to atherosclerotic processes [13], and recent studies indicate a role for proinflammatory lymphocytes infiltration in adipose tissue leading to insulin resistance [14]. Since there are few studies that have looked at the association of altered gene expression in Type 2 diabetes, and those that have, are all in European populations, we made an attempt to profile gene expression patterns in glucose intolerant Asian Indians, who are more prone for Type 2 diabetes and cardiovascular diseases.
Materials and methods
The study subjects were recruited from the Chennai Urban Rural Epidemiology Study (CURES), an ongoing epidemiological study conducted on a representative population (aged ≥20 years) of Chennai (formerly Madras), the fourth largest city in India. The methodology of the study has been published elsewhere [15]. In the study groups (n = 10 in each category), those who were confirmed by OGTT to have 2 h plasma glucose value ≥11.1 mmol/l [200 mg/dl] based on WHO consulting group criteria [15] were diagnosed as Type 2 diabetes patients, those with 2 h post-glucose value ≥7.8 mmol/l [140 mg/dl] and <11.1 mmol/l [200 mg/dl] [15] as IGT and those with 2 h post-glucose value <7.8 mmol/l [140 mg/dl] as normal glucose tolerance (NGT). All subjects were chosen randomly. IGT subjects were not on any medication. Of the diabetic patients, 9/10 were on oral hyperglycemic agents (OHA) and one had OHA plus insulin. The study has been carried out in accordance with the Declaration of Helsinki (2000) of the World Medical Association. Institutional ethical committee approval was obtained for the study and informed consent was obtained from all study subjects.
Anthropometric measurements
Anthropometric measurements including weight, height, and waist measurements were obtained using standardized techniques as detailed elsewhere [16]. Height was measured with a tape to the nearest cm. Weight was measured with traditional spring balance that was kept on a firm horizontal surface. Waist was measured using a non-stretchable fibre measure tape. The body mass index (BMI) was calculated using the formula, weight (kg)/height (m2). Blood pressure was recorded in the sitting position in the right arm to the nearest 2 mm Hg with a mercury sphygmomanometer (Diamond Deluxe BP apparatus, Pune, India). Two readings were taken 5 min apart and the mean of the two was taken as the blood pressure.
Biochemical parameters
Fasting plasma glucose (glucose oxidase-peroxidase method), serum cholesterol (cholesterol oxidase-peroxidase-amidopyrine method) serum triglycerides (glycerol phosphate oxidase-peroxidase-amidopyrine method), and HDL cholesterol (direct method-polyethylene glycol-pretreated enzymes) were measured using Hitachi-912 Autoanalyser (Hitachi, Mannheim, Germany). The intra- and inter-assay co-efficient of variation for the biochemical assays ranged between 3.1% and 7.6%. Low-density lipoprotein (LDL) cholesterol was calculated using the Friedewald formula [17]. Glycated haemoglobin (HbA1c) was estimated by high-pressure liquid chromatography using the Variant machine (Bio-Rad, Hercules, Calif., USA). The intra- and inter-assay co-efficient of variation of HbA1c was <10%.
Isolation of lymphocytes
Freshly collected peripheral blood was carefully layered on histopaque gradient (1,077) and centrifuged at 1,600 rpm (500 g) for 30 min. The buffy-coat interface representing >90% lymphocytes was aspirated and washed three times in phosphate-buffered saline (PBS) pH 7.4 [18, 19].
Gene expression studies
The gene expression profile was studied by semi-quantitative RT-PCR. Total RNA was extracted from lymphocytes by Triazol method (Sigma-Aldrich, USA) and the quality and quantity were confirmed by running an RNA gel. One microgram total RNA was used for the RT reaction and first-strand cDNA synthesis was carried out using random hexamers (Qiagen Inc., USA), MMLV reverse transcriptase according to manufacturer’s protocol. A reaction without reverse transcriptase was carried out as a negative control to ensure that the resulting DNA fragment was not due to amplification of contaminant DNA. The cDNA was amplified using human Beta-actin primers, forward-5′GGACTTCGAGCAAGAGATGG3′, reverse-5′AGAAAGGAAGGCTGGAAGA 3′. The PCR reaction protocol was run with an initial denaturation of 95°C for 5 min followed by 95°C for 1 min, 57°C for 1 min, 72°C for 1 min for 30 cycles with final elongation of 72°C for 10 min. Human TNF-α primers, forward-5′-GGCGGTGCTTGTTCCTCA -3′ and reverse-5′-TTCGAGAAGATGATCTGACTGCC-3′ [95°C for 30 s, 62°C for 30 s and 72°C for 30 s for 30 cycles], p22phox primers, forward-5′-GTTTGTGTGCCTGCTGGAG-3′ and reverse-5′-TGGGCGGCTGCTTGATGGT-3′ [95°C for 1 min, 62°C for 1 min, and 72°C for 1 min for 30 cycles], IL-6 primers, forward-5′-AGGAGACTTGCCTGGTGAAA-3′ and reverse-5′-CAGGGGTGGTTATTGCATCT-3′ [95°C for 30 s, 63°C for 30 s, and 72°C for 30 s for 30 cycles], TXNIP primers, forward-5′-CCACACCTTGGTCCTCATCT-3′ and reverse-5′-AGGAGGTGGAAGGGATGACT-3′ [95°C for 45 s, 59°C for 45 s, and 72°C for 45 s for 30 cycles], SOCS- 3 primers, forward-5′-GCCACCTACTGAACCCTCCT-3′ and reverse-5′-ACGGTCTTCCGACAGAGATG-3′ [95°C for 30 s, 59°C for 30 s, and 72°C for 30 s for 30 cycles]. The products were visualized in a 1.5% agarose gel and the bands were semi-quantified using gel documentation.
Protein carbonyls
Carbonyl content was evaluated by the 2,4-dinitrophenylhydrazine (DNPH) assay [20]. Blanks were run with 2 M HCl alone instead of DNPH reagent. Carbonyl content was expressed as nM/mg protein, using a molar absorption coefficient of 22,000 M−1 cm−1. Protein concentration was determined using a standard curve with bovine serum albumin (BSA: 0.25–5.0 mg/ml) dissolved in guanidine hydrochloride and read at 280 nm.
Lipid peroxidation
Plasma levels of malondialdehyde (MDA), a marker of lipid peroxidation, was measured by thiobarbituric acid reactive substances (TBARS) using a fluorescence methodology [21]. Briefly, 0.2 ml of plasma was mixed with 10% SDS, 20% acetic acid, and 0.53% TBA, and boiled at 100°C for 1 h. Butanol:pyridine (49:1) was added, mixed, and centrifuged at 3,000 rpm for 10 min. The organic solvent was removed and read at excitation 535 nm and emission 552 nm. Absolute MDA levels were calculated by regression parameters using different concentrations of the standard, 1,1′,3,3′-tetramethoxypropane.
Statistical analysis
Comparison between groups were performed using unpaired Student’s t-test and one-way ANOVA with P < 0.05 as the criterion for significance. Pearson correlation analysis was carried out to determine the relation of gene expression with other risk variables. Regression analysis was done to determine the association of gene expression with HbA1c. All analyses were done using Windows based SPSS statistical package (version 10.0, Chicago, IL).
Results
Table 1 shows the clinical and biochemical features of the study subjects. Subjects with glucose intolerance (i.e., IGT, Type 2 diabetic subjects) were older compared to subjects with NGT. Fasting plasma glucose, HbA1c, total cholesterol, and serum triglycerides were higher among subjects with IGT and diabetes compared to subjects with NGT (P < 0.001).
TBARS values were higher in subjects with IGT (0.44 nM/ml) compared to the subjects with NGT (0.37 nM/ml) and highest in those with diabetes (0.67 nM/ml, P < 0.05) (Fig. 1). PCO levels were also higher in subjects with diabetes (0.44 nmol/mg protein, P < 0.001) followed by IGT (0.21 nmol/mg protein) compared to NGT (0.17 nmol/mg protein) (Fig. 2).

Plasma TBARS levels among subjects with varying glucose tolerance. Data presented as mean and SEM. * P < 0.05 compared to NGT

Plasma protein carbonyl levels among subjects with varying glucose tolerance. Data presented as mean and SEM. * P < 0.01 compared to NGT. # P < 0.001 compared to IGT
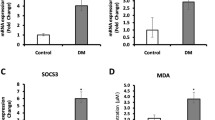
Figure 3a illustrates gene expression profiles of various genes. Both IL-6 (Fig. 3b) and TNF-α gene (Fig. 3c) expression (mean ± SE) were higher in diabetic subjects (IL-6: 0.80 ± 0.02, P < 0.01; TNF-α: 0.64 ± 0.04, P < 0.001) and in subjects with IGT (IL-6: 0.51 ± 0.04; TNF-α: 0.49 ± 0.06, P < 0.01) compared to normal subjects (IL-6: 0.28 ± 0.06, TNF-α: 0.19 ± 0.08). SOCS-3 gene expression was significantly decreased in subjects with diabetes (DM: 0.19 ± 0.02, P < 0.05) compared to IGT (0.33 ± 0.04) and NGT group (0.84 ± 0.05) (Fig. 3d). Subjects with diabetes and IGT had higher levels of NADPH oxidase (Fig. 3e) gene expression (DM: 1.50 ± 0.08, P < 0.05; IGT: 0.89 ± 0.06) compared to NGT group (0.49 ± 0.07). TXNIP (Fig. 3f) gene expression was also higher in those with diabetes and IGT (DM: 0.84 ± 0.07, P < 0.001; IGT: 0.58 ± 0.06) compared to NGT group (0.41 ± 0.07). The ‘P’ for trend was statistically significant (P < 0.01) for all the measured parameters. After age-adjustment, the trend for increased inflammatory and oxidative stress markers among subjects with IGT and patients with Type 2 diabetes persisted.

mRNA expression patterns in the study group. a Representative patterns of gene expression of IL-6, TNF-α, SOCS-3, NADPH oxidase [P22 Phox], TXNIP, and Beta Actin in control subjects (lanes 2–3), IGT subjects (lanes 4–5) and diabetic subjects (lanes 6–7). Lane 1 is the DNA ladder (50 bp). Densitometric analysis was carried out for the cumulative data on respective genes, IL-6 (b), TNF-α (c), SOCS-3 (d), NADPH oxidase [P22 Phox] (e), TXNIP (f) after adjusting for the expression of the housekeeping gene Beta Actin. * P < 0.05, ** P < 0.01 compared to NGT
Table 2 shows the Pearson correlation analysis between IL-6, TNF-α, NADPH oxidase, and TXNIP gene expression with metabolic risk factors. IL-6 gene expression showed a correlation with PCO (P < 0.001), fasting plasma glucose (P < 0.05), HbA1c (P < 0.01), TNF-α (P = 0.04), and NADPH oxidase (P = 0.02). TNF-α, showed a correlation with Tbars (P < 0.01), PCO (P < 0.001), fasting plasma glucose (P < 0.01), HbA1c (P < 0.001), gene expression of NADPH oxidase (P = 0.006), and TXNIP (P = 0.006). NADPH oxidase and TXNIP showed a positive correlation with fasting plasma glucose (P < 0.01) and HbA1c (P < 0.01). NADPH oxidase is positively correlated with Tbars (P < 0.05), PCO (P < 0.01), and TXNIP (P < 0.001). SOCS-3 was negatively correlated with PCO (P = 0.033), fasting plasma glucose (P = 0.035), HbA1c (P = 0.039), TNF-α (P = 0.022), NADPH oxidase (P = 0.04), and TXNIP (P = 0.033).
Multiple linear regression analysis was done using HbA1c as dependent variable and gene expression as independent variables. TXNIP and TNF-α showed a significant association with HbA1c even after adjusting for TBARS and PCO (TXNIP: β = 1.70, P < 0.01; TNF-α: β = 1.86, P < 0.01). NADPH Oxidase, IL-6 and SOCS-3 showed an association with HbA1c but this association was abolished after introducing TBARS and PCO into the model (Table 3).
Discussion
This study envisages three important findings. First, subjects with glucose intolerance exhibited increased proinflammatory gene expression along with compromised feedback regulation by SOCS signals. Secondly, these subjects were characterized by increased cellular pro-oxidation as revealed by increased gene expression of both NADPH oxidase and TXNIP. Thirdly and for the first time, these altered gene expression profiles were demonstrated to occur as early as in IGT.
The recent insight that inflammation contributes to the development of atherosclerosis and Type 2 diabetes mellitus constitutes a major breakthrough in understanding the mechanisms underlying these conditions. Chronic inflammatory conditions have been shown to be associated with a proatherogenic lipid pattern and altered glucose tolerance. Many studies showed that adipose tissue secretes inflammatory cytokines, which in turn contribute to IGT, insulin resistance, and Type 2 diabetes [22]. Circulating levels of cytokines, such as Tumor Necrosis Factor-α (TNF-α) and interleukin-6 (IL-6), and acute-phase proteins, such as C-reactive protein, are elevated in obesity, metabolic syndrome, and Type 2 diabetes [23, 24]. However, there are limited data on gene expression patterns of proinflammatory markers in clinical diabetes [25]. In this context, increased TNF-α and IL-6 gene expression pattern seen in subjects with IGT and Type 2 diabetes patients in this study is an important observation. Chronic inflammation in general and TNF-α in particular is likely to represent driving force for two events. First, TNF-α, as part of the inflammatory cascade originating from lymphocytes, might play a crucial role in the development of atherosclerotic lesions and able to induce proatherogenic lipoprotein changes. In this context, atherogenic TNF-α that are related to T cell function were demonstrated within atherosclerotic plaques [26, 27]. Secondly, TNF-α, by decreasing insulin sensitivity in adipose tissue might contribute to the development of glucose metabolism disturbances [28]. The later is supported by a recent study in which proinflammatory lymphocyte infiltration has been demonstrated as a primary event (prior to macrophage infiltration) in adipose tissue inflammation and the development of obesity-mediated insulin resistance [14].
In the Finnish diabetes prevention study, it has been shown that promoter polymorphisms of the TNF-α (G-308A) and IL-6 (C-174G) genes predict the conversion from IGT to Type 2 diabetes. In addition, a functional polymorphism of the TNF-α gene has been recently shown associated with Type 2 diabetes [29]. The −308A allele of the TNF-α gene has been found to increase TNF-α transcription [30] and secretion [31]. The progressive increase in TNF-α and IL-6 gene expression patterns in our study from subjects with IGT to Type 2 diabetes patients imply that these proinflammatory signals are important components of the risk factors of Type 2 diabetes and its complications. While the offspring of Type 2 diabetic subjects are at increased risk of diabetes, a recent study [32] showed that CRP and proinflammatory cytokine levels were elevated in nondiabetic offspring compared with the control group, supporting the concept that low-grade inflammation is one of the earliest findings in the pathogenesis of Type 2 diabetes.
Decreased SOCS-3 gene expression in subjects with glucose intolerance compared to normal glucose tolerant individuals is an important observation in this study. SOCS proteins regulate intensity and duration of cytokine/growth-factor signals and integrate multiple extracellular signals that may converge on target cells. SOCS-1 and SOCS-3 have been extensively studied both in vitro and in vivo in the context of insulin action. Interestingly, the level of SOCS-3 expression is strikingly enhanced in insulin-sensitive tissues from both patients and animal models with Type 2 diabetes and insulin resistance [33, 34]. Contrary to this, downregulation of SOCS-3 gene expression in glucose intolerant subjects observed by us could be a tissue-specific effect. It has been shown that SOCS-3 regulates proliferation and activation of lymphocytes [35] and counteracts uncontrolled activation of Th1 and Th2 responses. The gene expression pattern of SOCS-3 with both IL-6 and TNF-α in our study in control subjects supports this view. Thus, it appears that the physiological role of the SOCS proteins is most likely to prevent uncontrolled cytokine signaling in the cell by negative feedback. However, this mechanism seems to be compromised in glucose intolerant subjects as they exhibit low levels of SOCS-3, despite increased transcription of TNF-α and IL-6. It is evident from our study that the induction of SOCS expression is delayed or insufficient to overcome the destructive effect of the cytokines in glucose intolerant individuals. We suggest that SOCS proteins, particularly SOCS3, may regulate an important checkpoint that prevents inappropriate activation of lymphocyte subtypes or secretion of cytokines that underlie pathogenic mechanisms of Type 2 diabetes.
Increased NADPH oxidase activity is implicated in the pathogenesis of pancreatic β-cell apoptosis [36] and in the development of diabetic complications. Recently, it has been shown differential gene expression of NADPH oxidase (p22phox) and hemoxygenase-1 in patients with Type 2 diabetes and microangiopathy [37]. While our study supports this, it is important to note that increased pro-oxidant gene expression appear as early as in IGT.
Another important observation in this study is the progressively increased TXNIP gene expression in subjects with IGT and Type 2 diabetic patients. Chen et al. [38] have demonstrated that TXNIP induction plays a critical role in the vicious cycle of β-cell glucose toxicity and apoptosis. In addition, a recent human study [39] demonstrated that TXNIP modifies glucose uptake in adipocytes and skeletal muscle, and TXNIP expression was elevated even in the muscle of prediabetics. While TXNIP functions as an oxidative stress mediator by inhibiting thioredoxin activity, it has been shown to activate apoptotic (JNK) and proinflammatory signals [40]. Our study supports this as TXNIP gene expression is positively associated with protein oxidation and gene expression patterns of IL-6, TNF-α, and NADPH oxidase in patients with Type 2 diabetes.
Our study points out complex interactions among oxidative mediators in the prediabetic and hyperglycemic milieu. Elevations in TXNIP could represent a candidate intermediate linking diabetogenic stimuli to ROS production [41] and TNF-α stimuli to inflammation [42]. NADPH oxidase being a major source of ROS could elevate TXNIP whose induction is connected with an oxidative stress response. Alternatively, reduced levels of SOCS-3 might evoke hyperproduction of proinflammatory signals (TNF-α) which in turn augment oxidative stress. In our study, protein oxidation as measured by protein carbonyl content (PCO) was increased in subjects with IGT and Type 2 diabetes patients. Given that AOPPs (Advanced Oxidation Protein products) accumulation occurs from an early stage of prediabetes, when hyperglycemia was not present, it is plausible to propose that AOPPs accumulation resulting from oxidative stress in prediabetes can worsen the redox imbalance [43] and inflammation [44]. The enhanced oxidative stress and inflammation may further increase AOPPs formation via stimulation of lymphocytes to produce more oxidants. These proinflammatory/pro-oxidative lymphocyte phenotypes when infiltrate other organs might mediate atherogenesis [45], apoptosis [46], insulin resistance [14], and obesity [47]. Further studies should establish the role of specific lymphocyte subtypes in the evolution of metabolic and vascular dysfunction.
One of the limitations of this study is the small sample size and this was mostly related to matching the IGT subjects with other groups. Despite this, the study has given directionality for the role of immune cells in accelerating inflammation/oxidation which would have detrimental consequences in the target tissues of insulin signaling such as adipocytes. In addition, this study has also unraveled the role of certain novel drug targets such as SOCS-3 and TXNIP at the cross-road of prediabetes and Type 2 diabetes. To conclude, our study has demonstrated increased proinflammatory and pro-oxidant gene expression patterns not only in patients with Type 2 diabetes but also in subjects with IGT. We suggest that counteracting sub-clinical inflammation and/or oxidation should be one of the important strategies both in the treatment and prevention of Type 2 diabetes.
References
Gokulakrishnan K, Deepa R, Mohan V (2008) Association of high sensitivity C-reactive protein [hsCRP] and tumour necrosis factor-alpha [TNF-alpha] with carotid intimal medial thickness in subjects with different grades of glucose intolerance—the Chennai urban rural epidemiology study (CURES-31). Clin Biochem 41:480–485
Deepa R, Velmurugan K, Arvind K et al (2006) Serum levels of interleukin 6, C-reactive protein, vascular cell adhesion molecule 1, and monocyte chemotactic protein 1 in relation to insulin resistance and glucose intolerance—the Chennai urban rural epidemiology study (CURES). Metabolism 55:1232–1238. doi:10.1016/j.metabol.2006.05.008
Sampathkumar R, Balasubramanyam M, Sudarslal S, Rema M, Mohan V, Balaram P (2005) Increased glutathionylated hemoglobin (HbSSG) in type 2 diabetes subjects with microangiopathy. Clin Biochem 38:892–899. doi:10.1016/j.clinbiochem.2005.06.009
Adaikalakoteswari A, Balasubramanyam M, Mohan V (2005) Telomere shortening occurs in Asian Indian type 2 diabetic patients. Diabet Med 22:1151–1156. doi:10.1111/j.1464-5491.2005.01574.x
Knowler WC, Narayan KM, Hanson RL et al (1995) Preventing non-insulin-dependent diabetes. Diabetes 44:483–488. doi:10.2337/diabetes.44.5.483
Gokulakrishnan K, Deepa R, Velmurugan K, Ravikumar R, Karkuzhali K, Mohan V (2007) Oxidized low-density lipoprotein and intimal medial thickness in subjects with glucose intolerance: the Chennai urban rural epidemiology study-25. Metabolism 56:245–250. doi:10.1016/j.metabol.2006.10.002
Gokulakrishnan K, Deepa R, Mohan V, Gross MD (2006) Soluble P-selectin and CD40L levels in subjects with prediabetes, diabetes mellitus, and metabolic syndrome—the Chennai urban rural epidemiology study. Metabolism 55:237–242. doi:10.1016/j.metabol.2005.08.019
Donahue RP, Rejman K, Rafalson LB, Dmochowski J, Stranges S, Trevisan M (2007) Sex differences in endothelial function markers before conversion to pre-diabetes: does the clock start ticking earlier among women? the Western New York study. Diabetes Care 30:354–359. doi:10.2337/dc06-1772
Patti ME (2004) Gene expression in humans with diabetes and prediabetes: what have we learned about diabetes pathophysiology? Curr Opin Clin Nutr Metab Care 7:383–390. doi:10.1097/01.mco.0000134359.23288.72
Balasubramanyam M, Premanand C, Mohan V (2002) The lymphocyte as a cellular model to study insights into the pathophysiology of diabetes and its complications. Ann NY Acad Sci 958:399–402
Srinivasan V, Sandhya N, Sampathkumar R, Farooq S, Mohan V, Balasubramanyam M (2007) Glutamine fructose-6-phosphate amidotransferase (GFAT) gene expression and activity in patients with type 2 diabetes: inter-relationships with hyperglycaemia and oxidative stress. Clin Biochem 40:952–957. doi:10.1016/j.clinbiochem.2007.05.002
Stentz FB, Kitabchi AE (2007) Transcriptome and proteome expressions involved in insulin resistance in muscle and activated T-lymphocytes of patients with type 2 diabetes. Genomics Proteomics Bioinformatics 5:216–235. doi:10.1016/S1672-0229(08)60009-1
Robertson AK, Hansson GK (2006) T cells in atherogenesis: for better or for worse? Arterioscler Thromb Vasc Biol 26:2421–2432. doi:10.1161/01.ATV.0000245830.29764.84
Kintscher U, Hartge M, Hess K et al (2008) T-lymphocyte infiltration in visceral adipose tissue: a primary event in adipose tissue inflammation and the development of obesity-mediated insulin resistance. Arterioscler Thromb Vasc Biol 28:1304–1310. doi:10.1161/ATVBAHA.108.165100
Deepa M, Pradeepa R, Rema M et al (2003) The Chennai urban rural epidemiology study (CURES)—study design and methodology (urban component) (CURES-I). J Assoc Physicians India 51:863–870
Alberti KG, Zimmet PZ (1998) Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: Diagnosis and classification of diabetes mellitus, provisional report of a WHO Consultation. Diabet Med 15:539–553. doi:10.1002/(SICI)1096-9136(199807)15:7<539::AID-DIA668>3.0.CO;2-S
Friedewald WT, Levy RI, Fredrickson DS (1972) Estimation of the concentration of low-density lipoprotein cholesterol in plasma without use of the preparative ultracentrifuge. Clin Chem 18:499–502
Balasubramanyam M, Kimura M, Aviv A, Gardner JP (1993) Kinetics of calcium transport across the lymphocyte plasma membrane. Am J Physiol 265:C321–C327
Balasubramanyam M, Rohowsky-Kochan C, Reeves JP, Gardner JP (1994) Na+/Ca2+exchange-mediated calcium entry in human lymphocytes. J Clin Invest 94:2002–2008. doi:10.1172/JCI117553
Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz AG (1990) Determination of carbonyl content in oxidatively modified proteins. Methods Enzymol 186:464–478. doi:10.1016/0076-6879(90)86141-H
Yagi K (1976) A simple fluorometric assay for lipoperoxide in blood plasma. Biochem Med 15:212–216. doi:10.1016/0006-2944(76)90049-1
Gimeno RE, Klaman LD (2005) Adipose tissue as an active endocrine organ: recent advances. Curr Opin Pharmacol 5:122–128. doi:10.1016/j.coph.2005.01.006
Pickup JC, Mattock MB, Chusney GD, Burt D (1997) NIDDM as a disease of the innate immune system: association of acute-phase reactants and interleukin-6 with metabolic syndrome X. Diabetologia 40:1286–1292. doi:10.1007/s001250050822
Mohan V, Deepa R, Velmurugan K, Premalatha G (2005) Association of C-reactive protein with body fat, diabetes and coronary artery disease in Asian Indians: the Chennai urban rural epidemiology study (CURES-6). Diabet Med 22:863–870. doi:10.1111/j.1464-5491.2005.01541.x
Tsiotra PC, Tsigos C, Yfanti E et al (2007) Visfatin, TNF-alpha and IL-6 mRNA expression is increased in mononuclear cells from type 2 diabetic women. Horm Metab Res 39:758–763. doi:10.1055/s-2007-990288
Branen L, Hovgaard L, Nitulescu M, Bengtsson E, Nilsson J, Jovinge S (2004) Inhibition of tumor necrosis factor-alpha reduces atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol 24:2137–2142. doi:10.1161/01.ATV.0000143933.20616.1b
Sato K, Niessner A, Kopecky SL, Frye RL, Goronzy JJ, Weyand CM (2006) TRAIL-expressing T cells induce apoptosis of vascular smooth muscle cells in the atherosclerotic plaque. J Exp Med 203:239–250. doi:10.1084/jem.20051062
Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM (1995) Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest 95:2409–2415. doi:10.1172/JCI117936
Susa S, Daimon M, Sakabe J et al (2008) A functional polymorphism of the TNF-alpha gene that is associated with type 2 DM. Biochem Biophys Res Commun 369:943–947. doi:10.1016/j.bbrc.2008.02.121
Wilson AG, Symons JA, McDowell TL, McDevitt HO, Duff GW (1997) Effects of a polymorphism in the human tumor necrosis factor alpha promoter on transcriptional activation. Proc Natl Acad Sci USA 94:3195–3199. doi:10.1073/pnas.94.7.3195
Bouma G, Crusius JB, Oudkerk Pool M et al (1996) Secretion of tumour necrosis factor alpha and lymphotoxin alpha in relation to polymorphisms in the TNF genes and HLA-DR alleles. Relevance for inflammatory bowel disease. Scand J Immunol 43:456–463. doi:10.1046/j.1365-3083.1996.d01-65.x
Ruotsalainen E, Salmenniemi U, Vauhkonen I et al (2006) Changes in inflammatory cytokines are related to impaired glucose tolerance in offspring of type 2 diabetic subjects. Diabetes Care 29:2714–2720. doi:10.2337/dc06-0147
Rui L, Yuan M, Frantz D, Shoelson S, White MF (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277:42394–42398. doi:10.1074/jbc.C200444200
Lebrun P, Van Obberghen E (2008) SOCS proteins causing trouble in insulin action. Acta Physiol (Oxf) 192:29–36
Yu CR, Mahdi RM, Ebong S, Vistica BP, Gery I, Egwuagu CE (2003) Suppressor of cytokine signaling 3 regulates proliferation and activation of T-helper cells. J Biol Chem 278:29752–29759. doi:10.1074/jbc.M300489200
Oliveira HR, Verlengia R, Carvalho CR, Britto LR, Curi R, Carpinelli AR (2003) Pancreatic β-cells express phagocyte-like NAD(P). Hoxidase. Diabetes 52:1457–1463. doi:10.2337/diabetes.52.6.1457
Adaikalakoteswari A, Balasubramanyam M, Rema M, Mohan V (2006) Differential gene expression of NADPH oxidase (p22phox) and hemoxygenase-1 in patients with Type 2 diabetes and microangiopathy. Diabet Med 23:666–674. doi:10.1111/j.1464-5491.2006.01879.x
Chen J, Saxena G, Mungrue IN, Lusis AJ, Shalev A (2008) Thioredoxin-interacting protein: a critical link between glucose toxicity and beta-cell apoptosis. Diabetes 57:938–944. doi:10.2337/db07-0715
Parikh H, Carlsson E, Chutkow WA et al (2007) TXNIP regulates peripheral glucose metabolism in humans. PLoS Med 4:e158. doi:10.1371/journal.pmed.0040158
Junn E, Han SH, Im JY et al (2000) Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J Immunol 164:6287–6295
Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT (2004) Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem 279:30369–30374. doi:10.1074/jbc.M400549200
Yamawaki H, Pan S, Lee RT, Berk BC (2005) Fluid shear stress inhibits vascular inflammation by decreasing thioredoxin-interacting protein in endothelial cells. J Clin Invest 115:733–738
Wei XF, Zhou QG, Hou FF, Liu BY, Liang M (2008) Advanced oxidation protein products induce mesangial cells perturbation through PKC-dependent activation of NADPH oxidase. Am J Physiol Renal Physiol. doi:10.1152/ajprenal.90536.2008
Shi XY, Hou FF, Niu HX et al (2008) Advanced oxidation protein products promote inflammation in diabetic kidney through activation of renal nicotinamide adenine dinucleotide phosphate oxidase. Endocrinology 149:1829–1839. doi:10.1210/en.2007-1544
Hansson GK, Holm J, Jonasson L (1989) Detection of activated T lymphocytes in the human atherosclerotic plaque. Am J Pathol 135:169–175
Hänninen A, Nurmela R, Maksimow M, Heino J, Jalkanen S, Kurts C (2007) Islet beta-cell-specific T cells can use different homing mechanisms to infiltrate and destroy pancreatic islets. Am J Pathol 170:240–250. doi:10.2353/ajpath.2007.060142
Rocha VZ, Folco EJ, Sukhova G, Shimizu K, Gotsman I, Vernon AH, Libby P (2008) Interferon-gamma, a Th1 cytokine, regulates fat inflammation: a role for adaptive immunity in obesity. Circ Res 103:467–476. doi:10.1161/CIRCRESAHA.108.177105
Acknowledgements
This work was supported by research grants from the Department of Science and Technology (DST-FIST), Indian Council of Medical Research (ICMR) and the Department of Biotechnology (DBT), Government of India, New Delhi, India. KG and FM acknowledges the financial assistance (Senior Research Fellowship) from the Council of Scientific & Industrial Research (CSIR), New Delhi, India. This is paper no. 49 from the Chennai Urban Rural Epidemiology Study (CURES).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gokulakrishnan, K., Mohanavalli, K.T., Monickaraj, F. et al. Subclinical inflammation/oxidation as revealed by altered gene expression profiles in subjects with impaired glucose tolerance and Type 2 diabetes patients. Mol Cell Biochem 324, 173–181 (2009). https://doi.org/10.1007/s11010-008-9996-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-008-9996-x