
Abstract
Oxidative stress is important in the pathogenesis of renal ischemia-reperfusion (IR) injury; however whether imbalances in reactive oxygen production and disposal account for susceptibility to injury is unclear. The purpose of this study was to compare necrosis, apoptosis, and oxidative stress in IR-resistant Brown Norway rats vs. IR-susceptible Sprague-Dawley (SD) rats in an in vivo model of renal IR injury. As superoxide (O ·−2 ) interacts with nitric oxide (NO) to form peroxynitrite, inducible NO synthase (iNOS) and nitrotyrosine were also examined. Renal IR was induced in SD and BN rats by bilateral clamping of renal arteries for 45 min followed by reperfusion for 24 h (SD 24 and BN 24, respectively). BN rats were resistant to renal IR injury as evidenced by lower plasma creatinine and decreased acute tubular necrosis. TUNEL staining analysis demonstrated significantly decreased apoptosis in the BN rats vs. SD rats after IR. Following IR, O ·−2 levels were also significantly lower in renal tissue of BN rats vs. SD rats (P < 0.05) in conjunction with a preservation of the O ·−2 dismutating protein, CuZn superoxide dismutase (CuZn SOD) (P < 0.05). This was accompanied by an overall decrease in 4-hydroxynonenal adducts in the BN but not SD rats after IR. BN rats also displayed lower iNOS expression (P < 0.05) resulting in lower tissue NO levels and decreased nitrotyrosine formation (P < 0.01) following IR. Collectively these results show that the resistance of the BN rat to renal IR injury is associated with a favorable balance of oxidant production vs. oxidant removal.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Acute renal failure, most commonly induced by ischemia-reperfusion (IR), affects as many as 5% of all hospitalized patients and has an unacceptably high rate of mortality [1–3]. Currently there are no specific therapies available to prevent or improve recovery in acute renal failure. While much attention has been focused on the mechanisms by which IR causes renal failure, little is known about how the kidney might be protected from IR injury. Elucidation of mechanisms of intrinsic resistance to injury provides the opportunity to dissect the relative importance of putative protective pathways on the pathogenesis of acute renal failure.
Oxygen-derived free radicals have been implicated in the pathogenesis of IR-induced renal injury [4]. During reperfusion, the conversion of accumulated hypoxanthine to xanthine generates O ·−2 [5, 6]. Concomittantly, ischemia induces nitric oxide synthase (iNOS) in tubule cells generating nitric oxide (NO). While some studies have shown that NO is protective against free radical-mediated injury [7–9] and diminishes apoptosis after renal IR [10], other investigators have demonstrated a damaging role for iNOS in IR [11, 12]. It is, however, generally believed that NO can combine with O ·−2 to form the potently nitrating and damaging species, peroxynitrite [13–17].
The major cellular mechanism for eliminating reactive oxygen species is the antioxidant enzyme system that includes the superoxide dismutases (CuZn SOD and Mn SOD) that convert O ·−2 to H2O2 and enzymes (catalase and glutathione peroxidase) that degrade H2O2 to H2O. Paradoxically, IR results in impairment of intrinsic cellular antioxidant defenses, which in turn renders the cell more susceptible to oxidative stress [18–20]. Specifically, it has been demonstrated that the major mitochondrial antioxidant enzyme, manganese superoxide dismutase (Mn SOD), is inactivated following warm ischemia in rat kidneys [21]. Further, the cytoplasmic form of the protein, copper zinc superoxide dismutase (CuZn SOD) is rapidly depleted following warm ischemia [18]. Over-expression of either SOD isoform results in protection from ATP depletion or renal IR injury [22, 23] further demonstrating the importance of the O ·−2 removing enzymes in mitigating IR-induced oxidative stress.
Recently, the BN rat strain was discovered to be dramatically resistant to cardiac and renal IR injury compared with other rat strains [24–26]. The protection from renal IR injury was demonstrated early by maintenance of Na+-K+ ATPase distribution on the cellular levels and later by inhibition of increases in creatinine concentration, lack of changes in morphological structure and the maintenance of control-level excretion rates of Na and water [25]. While one potential contribution to this resistance was defined, the mechanisms leading to this resistance have not been delineated. Recent investigations in a cardiac IR model demonstrated that BN/Mcwi rats generated less O ·−2 , by maintenance of endothelial NOS function [26] compared to Dahl S (SS/Mcwi) rats, suggesting that increases in O ·−2 may be responsible for the dysfunction in this model. Thus, using the BN rats to determine whether specific pathways of injury/protection are altered can give mechanistic insights into which pathways are involved in resistance/susceptibility to renal IR injury.
In this study, we hypothesized that the observed resistance in BN rats to IR-induced acute renal failure may be mediated through altered renal O ·−2 - and NO-generating systems and O ·−2 -dismutating enzymes. To explore this possibility, we compared apoptosis and oxidative/nitrative stress parameters in SD rats and BN rats in an in vivo model of IR injury.
Methods
Animal model of renal IR injury
SD rats and BN rats, purchased from Harlan (Harlan, Indianapolis, IN) were used. All experiments were approved by the institutional Animal Care and Use committee at the Zablocki VA Medical Center. We used a model of IR injury previously established by one of the co-authors [25]. Renal ischemia was induced in BN and SD rats (BN 24, SD 24) by bilateral clamping of the renal arteries for 45 min after anesthetizing animals with sodium pentobarbital (50 mg/kg i.p.). Reperfusion was verified by visual inspection following removal of the microvascular clips. Sham controls underwent surgery without renal artery clamping (BN sham, SD sham). After 24 h recovery, animals were anesthetized, blood samples obtained and kidneys isolated and quick frozen in liquid nitrogen. Kidneys were stored at −80°C until further analysis. Blood was sampled using heparinized syringes, centrifuged for 10 min at 1,500 rpm and plasma stored at −80°C until further analysis.
Measurement of plasma creatinine
Creatinine was measured in plasma from sham and IR rats using a commercial kit (Teco Diagnostics, Anaheim, CA). This method is based on the reaction of creatinine with picric acid to form a color complex which absorbs at 510 nm.
Histological scoring for acute tubular necrosis
Tissue from a portion of the kidney was fixed in 4% phosphate-buffered formalin (pH 7.4), and paraffin-embedded sections were stained with hemotoxylin and eosin (H&E) or periodic acid Schiff reagent (PAS). Histological scoring was performed in a blinded fashion and was based on a score ranging from 1 to 4, with 1 indicating minimal change and 4 indicating complete necrosis [27].
TUNEL staining for apoptosis
Apoptosis was measured by the TUNEL assay using ApopTag® technology (Chemicon International, Temecula, CA) according to manufacturer’s instructions. In this method, DNA strand breaks are detected by enzymatically labeling the free 3′-OH termini with modified nucleotides with terminal deoxynucleotidyl transferase (TdT). DNA fragments are then labeled with digoxigenin and bound to an anti-digoxigenin antibody conjugated to horseradish peroxidase. The peroxidase conjugate produces a localized stain (apoptotic body) which can be detected and counted. Sections were counterstained with H&E. A minimum of three sections per slide and five fields per section were counted and apoptotic nuclei quantified.
RNA levels
Total RNA was purified from approximately 60 mg of frozen tissue using the Promega SV RNA total RNA isolation kit (Promega, Madison, WI). One μg RNA was reverse transcribed using the Invitrogen Superscript First Strand Synthesis System (Invitrogen, Carlsbad, CA) with oligo (dT) primers as previously described [28]. Amplification of rat iNOS, CuZn SOD, and Mn SOD was performed using the following primers: 5′-caccttggagttcacccagt-3′ (sense; iNOS), 5′-tgttgtagcgctgtggtca-3′ (antisense; iNOS), 5′-ggagagcattccatcattgg-3′ (sense; CuZn SOD), 5′-caatcacaccacaagccaag-3′ (antisense; CuZn SOD), and 5′-attaacgcgcagatcatgca-3′ (sense; Mn SOD), 5′-cctcggtgacgttcagattgt-3′ (antisense; Mn SOD). For PCR, 1 μl of cDNA was mixed with 25 pmol of each primer and PCR Supermix (Invitrogen, Carlsbad, CA) to a volume of 25 μl and incubated in an Applied Biosystems Gene Amp® PCR System 9700 (Applied Biosystems, Foster City, CA) under the following conditions: 94°C for 5 min, 94°C for 1 min, 60°C for 1 min, 72°C for 1 min for 30 cycles (iNOS), 95°C for 5 min, 95°C for 1 min, 60°C for 1 min, 72°C for 1 min (CuZn SOD), and 94°C for 5 min, 94°C for 1 min, 60°C for 45 s, 72°C for 45 s for 30 cycles (Mn SOD). The PCR products were resolved on a 1% agarose gel and visualized with ethidium bromide staining followed by densitometry. Rat GAPDH was used as a control for cDNA loading.
Lucigenin-enhanced chemiluminescence for O ·−2 detection
Approximately 100 mg of kidney tissue was homogenized in 10 ml of Krebs–Henseleit buffer containing 10 mM HEPES (pH = 7.4) similar to that previously described for cardiac tissue [29]. The tubes containing 1 ml of the homogenate or buffer as blank were incubated in the dark either alone or with 100 μM of the SOD mimetic, manganese (III) tetrakis (1-methyl-4-pyridyl) porphyrin pentachloride (MnTmPyP) for 30 min to test the specificity of the signal. Lucigenin-enhanced chemiluminescence was then measured for 3 min in a luminometer (Berthold Instruments, Autolomat LB 953, Bad Wilblad, Germany) after the addition of 10 μM lucigenin to each tube. The tube containing the buffer and lucigenin was read first to calculate background luminescence and this value was subtracted from each subsequent value. A BIO-RAD protein assay was performed for assessment of protein in mg/ml. Reactive oxygen production was reported as relative light units emitted (RLU)/mg protein.
Western blotting
Frozen tissue was homogenized in ice-cold, phosphate-buffered saline (PBS, pH = 7.4) with 1% Triton X-100, 1 mmol/l phenylmethylsulfonyl fluoride, 10 ng/ml each of pepstatin, leupeptin, and aprotinin. Homogenates were centrifuged at 10,000 × g for 10 min at 4°C. Protein concentration of the supernatant was determined by the Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA). Thirty μg of the protein was electrophoresed on either 7.5% or 12% SDS-polyacrylamide gels (for iNOS or CuZn SOD and Mn SOD, respectively) and transferred to Nytran membranes as described [30]. For determination of 4-hydroxy-2-nonenal adducts (HNE, a lipid peroxidation product), 10 μg of protein was electrophoresed on 12% SDS-polyacrylamide gels. After destaining of membranes and blocking in PBS-0.05% Tween 20 (1 h), blots were probed with one of the following antibodies: rabbit anti-iNOS (1:1,000, overnight at 4°C, Santa Cruz Biotechnology, CA), goat anti-CuZn SOD (1:1,000, 1 h at room temperature, Calbiochem, San Diego, CA), rabbit anti-MnSOD (1:1,000, 1 h at room temperature, Upstate Biotechnology, Lake Placid, NY), and rabbit anti-HNE (1:1,000, overnight at 4°C, Calbiochem, San Diego, CA) and visualized with a 1:5,000 or 1:10,000 dilution of IgG conjugated to horseradish peroxidase and enhanced chemiluminescence according to manufacturer’s instructions. Densitometry was performed on an AlphaImager 2000 Image Analysis System (Alpha Innotech, San Leandro, CA) and normalized to β-actin. Membranes were stripped using Restore™ Western blot stripping buffer at 37°C for 20 min and reprobed with goat anti-β-actin (1:1,000, overnight at 4°C, Upstate Biotechnology, Lake Placid, NY) as a loading control.
Nitrate/nitrite levels
Plasma NO by-products, nitrate and nitrite, were measured by a commercial kit (Cayman Chemical, Ann Arbor, MI) according to manufacturer’s guidelines. For tissue NO metabolite levels, tissues (∼100 mg) were homogenized in PBS (pH 7.4) and centrifuged at 10,000 × g for 20 min to remove cellular debris. The supernatant was ultracentrifuged at 100,000 × g for 30 min and filtrated with 10,000 MW cut-off filters (Amicon, Millipore Corporation, Billerica, MA). The resulting ultrafiltrate was used for the measurement of nitrate and nitrite using a commercial kit (Cayman Chemical, Ann Arbor, MI) using manufacturer’s guidelines. Protein concentration was determined by the Bio-Rad DC Protein Assay (Bio-Rad, Hercules CA).
Slot blot for nitrotyrosine
Slot-blot immunoassay was used for nitrotyrosine content as described [31]. Briefly, an aliquot of 3 μg of protein from kidney homogenates was applied to a Bio-Dot apparatus (Bio-Rad, Hercules, CA) in duplicate to nitrocellulose membranes. After blocking with 3% non-fat milk in PBS-0.05% Tween-20 for 1 h, the membrane was incubated overnight at 4°C with 0.8 μg/ml monoclonal anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY). The blot was visualized with anti-mouse IgG horseradish peroxidase and enhanced chemiluminescence. Serial dilutions of nitrated bovine serum albumin (BSA) standard were used to quantify nitrotyrosine.
Immunohistochemistry for nitrotyrosine
Immunohistochemical analysis for nitrotyrosine was performed as previously described with some modifications [31]. Briefly, a portion of kidney tissue was fixed and paraffin-embedded for immunohistochemical analysis. To quench endogenous background, sections were incubated with 1% H2O2 in methanol for 5 min and washed twice in 7.4% buffered saline. Non-specific binding of secondary antibody was blocked with BSA for 1 h before incubation with primary antibody. Sections were incubated with 1:50 dilution of monoclonal anti-nitrotyrosine as primary (Upstate Biotechnology, Lake Placid, NY) and horseradish peroxidase-conjugated secondary IgG (Upstate Biotechnology, Lake Placid, NY) and counter stained with methyl green. Negative controls included secondary antibody incubation alone without primary antibody (data not shown). For specificity, the positive control used was human tumor tissue purchased from Dako (Carpinteria, CA).
Statistical analyses
Results are expressed as mean ± SEM. Statistical analysis was done by Analysis of Variance (ANOVA) followed by Newman Keuls multiple comparison tests, with P-value set at 0.05.
Results
Kidney function, histological damage, and apoptosis
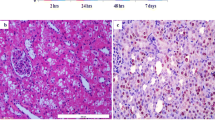
Plasma creatinine levels (for assessing kidney function) were increased significantly (P < 0.001) after renal IR in SD rats compared to BN or sham controls (Fig. 1A). Histological analysis of the kidney also revealed increased tubular necrosis in the SD rats vs. BN rats following IR (Fig. 1B). Histological scoring was based on a score ranging from 1 to 4, with 1 indicating minimal change, 2 indicating focal ATN, 3 indicating patchy to extensive ATN, and 4 indicating complete necrosis [27]. SD rats had damaged proximal tubular epithelium, cytoplasmic and basement membrane disruption, and exposed nuclei after IR compared to the BN rat (Fig. 1C). Apoptotic nuclei as measured by TUNEL staining were infrequent to absent in tissue sections from both BN and SD sham controls (Fig. 2). After IR, apoptotic nuclei increased significantly in kidneys of SD rats (Fig .2; P < 0.01, SD 24 vs. all other groups) but not in kidneys of the BN rats.

(A) Plasma creatinine levels in BN and SD sham and BN and SD rats following renal IR. Groups include BN sham, SD sham, BN 24, and SD 24. Results are means ± SEM. ¶ P < 0.001 SD vs. all other groups (n = 11 for BN 24 and n = 12 for BN sham, SD sham and SD 24). (B) Histological scoring showing acute tubular necrosis (ATN) in BN and SD rats following renal IR. Results are means ± SEM. (C) Representative image of PAS stained slide showing evidence of histological damage in SD 24 rats compared to BN sham, SD sham, and BN 24 rats. Arrows indicate disruption of basement membrane and exposed nuclei

Upper panel: TUNEL staining depicting apoptotic nuclei in SD sham, SD 24, BN sham, and BN 24 rats. Lower panel: Graph depicting apoptotic nuclei/high powered field in BN and SD rats after renal IR injury. Results are means ± SEM (n = 4 for all groups). ‡ P < 0.01 SD 24 vs. all other groups (n = 4 for all groups)
Superoxide levels and SOD expression
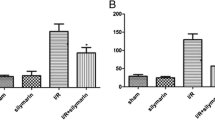
We next examined renal lucigenin-enhanced chemiluminescence for O ·−2 levels in BN and SD rats after IR. Lucigenin-enhanced chemiluminescence was slightly higher in kidneys of BN sham rats vs. SD sham rats; however, after IR, O ·−2 was dramatically increased in renal tissue of SD rats only (Fig. 3; P < 0.01 SD 24 vs. SD sham and BN sham; P < 0.05 SD 24 vs. BN 24). The signal was specific for O ·−2 , since treatment of the extract with 100 μM MnTmPyP, a SOD mimetic for 30 min completely abolished the signal (data not shown).

Lucigenin-enhanced chemiluminescence showing increased superoxide levels in SD 24 rats compared to BN sham, SD sham, and BN 24 rats. Results are means ± SEM. ¶ P < 0.001 SD 24 vs. SD sham, *P < 0.05 SD 24 vs. BN sham and BN 24 (n = 3 for all groups)
Western blot analysis revealed no change in MnSOD or CuZn SOD protein in BN rats after IR compared to BN sham controls (Fig. 4A). There was a slight decrease in the MnSOD in the SD 24 rats vs. BN 24 or sham controls after IR (Fig. 4A), although this difference did not reach statistical significance. CuZn SOD protein levels were unchanged after IR in BN rats compared to BN sham controls. In contrast, after IR, CuZn SOD was significantly decreased in SD 24 rats but not in BN 24 rats (Fig. 4A; P < 0.01, SD 24 vs. all other groups).

(A) Upper panel: Representative Western blot showing CuZn SOD and MnSOD protein levels in sham and reperfused BN and SD rats. Lower panel: Densitometry of CuZn SOD and MnSOD as normalized to β-actin in SD sham, SD 24, BN sham, and BN 24 rats. Results are means ± SEM *P < 0.05 SD 24 vs. all other groups (n = 11 for BN sham and SD sham, n = 12 for BN 24 and SD 24). (B) Upper panel: Representative reverse transcriptase PCR (RT-PCR) showing expression of Mn SOD and CuZn SOD relative to GAPDH mRNA in SD sham, SD 24, BN sham, and BN 24 rats. Lower panel: Densitometry of RT-PCR for CuZn SOD and Mn SOD expressed as a ratio to GAPDH showing a decrease in CuZn SOD and Mn SOD mRNA in SD 24 rats compared to BN 24 or BN and SD sham animals. Results are means ± SEM. *P < 0.05 SD 24 vs. SD sham (CuZn SOD, n = 3 for all groups); *P < 0.05 SD 24 vs. BN 24, SD sham and BN sham (MnSOD, n = 3 for all groups)
In order to determine if the decrease in SOD protein was due to transcriptional regulation, we did RT-PCR of Mn SOD and CuZn SOD in SD sham, SD 24, BN sham, and BN 24 animals. While there was no change in Mn SOD or CuZn SOD RNA in BN 24 rats compared to BN sham controls, there was a marked decrease in Mn SOD (Fig. 4B; P < 0.05 SD 24 vs. all other groups) and CuZn SOD RNA in the SD 24 rats (Fig. 4B; P < 0.05 SD 24 vs. SD sham) compared to SD sham control animals. This indicated that there is differential transcriptional regulation of the SOD proteins in the two strains after renal IR.
Lipid peroxidation
Oxidation of ω6-unsaturated fatty acids by superoxide releases HNE, a highly reactive and cytotoxic aldehyde that can initiate lipid peroxidation. HNE-protein adducts were measured to determine the extent of lipid peroxidation in BN and SD rats in sham controls and BN and SD animals subjected to renal IR. Western blotting indicated that there was low levels of HNE-protein adducts in the BN and SD sham controls (Fig. 5). Quantification of all the immunoreactive bands revealed that following IR, immunoreactive HNE was significantly increased preferentially in the SD rats compared to BN 24 rats or sham controls (Fig. 5; P < 0.001, SD 24 vs. SD sham and BN sham). While there was some increase (not statistically significant against corresponding sham controls) in HNE-adduct formation in the BN rats following IR this was significantly lower than the SD 24 rats (Fig. 5; P < 0.01 SD 24 vs. BN 24).

Upper panel: Western blot for HNE-protein adducts showing increased lipid peroxidation in SD rats (SD 24) after IR compared to the BN rats (BN 24) and respective sham groups. Lower panel: Densitometry of Western blot in SD sham, SD 24, BN sham, and BN 24 rats. Results are means ± SEM. (n = 3 for all groups). ¶ P < 0.001 SD 24 vs. SD sham and BN sham, ‡ P < 0.01 SD 24 vs. BN 24
iNOS protein, NO levels, and protein nitration
Using iNOS inhibitors and iNOS knockout mice, previous investigators have elucidated the importance of iNOS-derived NO in renal IR injury [11, 12]. We therefore examined if there was a difference in iNOS expression in BN and SD rats following renal IR injury. Both iNOS RNA (Fig. 6A) and protein (Fig. 6B) were up-regulated in the SD strain after IR compared to SD sham controls (Fig. 6B; P < 0.01, SD 24 vs. all other groups). However, iNOS RNA and protein was not increased in renal tissue of the BN rat after IR relative to either BN sham controls or IR in SD rats (Fig. 6A, B).

(A) Upper panel: RT-PCR showing iNOS mRNA relative to GAPDH mRNA in SD sham, SD 24, BN sham, and BN 24 rats. Lower panel: Densitometry of RT-PCR for iNOS expressed as a ratio to the housekeeping gene GAPDH showing an increase of iNOS mRNA in SD 24 rats compared to BN 24 or sham animals. Results are means ± SEM. Results are not significantly different from each other. (B) Upper panel: Representative Western blot showing iNOS protein levels in sham and reperfused BN and SD rats. Lower panel: Densitometry of iNOS as normalized to β-actin in SD sham, SD 24, BN sham, and BN 24 rats. Results are means ± SEM. ‡ P < 0.01 SD 24 vs. all other groups (n = 9 for all groups)
In order to determine whether increased iNOS expression translated to an increase in NO metabolite levels were measured in both plasma and renal tissue of BN sham, SD sham, BN 24, and SD 24 rats. Plasma NO metabolite levels were similar in both BN and SD sham rats (Fig. 7, upper panel). Interestingly, after IR, plasma NO levels were increased to a similar degree in both SD and BN groups (Fig. 7, upper panel; P < 0.01 BN 24 and SD 24 vs. respective sham controls). However, the pattern for tissue nitrate–nitrite levels was different. There was no difference in tissue NO metabolite levels in BN sham or SD sham rats (Fig. 7, lower panel). Following IR, renal tissue NO metabolite levels were increased in SD rats but not in BN rats, although this increase did not reach statistical significance (Fig. 7, lower panel; P = 0.075 SD 24 vs. BN 24).

Upper panel: Plasma nitrate + nitrite levels in SD sham, SD 24, BN sham, and BN 24 rats. Results are means ± SEM (n = 6–9 for all groups). ‡ P < 0.01 SD 24 and BN 24 vs. respective shams. Lower panel: Tissue nitrate + nitrite levels in sham and reperfused BN and SD animals. Results are means ± SEM. Results not statistically different from each other
O ·−2 and NO can interact and lead to the formation of the potent tyrosine nitrating species, peroxynitrite. Total protein nitrotyrosine was evaluated by slot-blot analysis in kidney homogenates from BN 24, SD 24, and BN and SD sham controls. Total nitrotyrosine protein levels in renal tissue were similar in time-matched, BN and SD surgery sham control rats. Following IR, there was a significant increase of total nitrotyrosine protein in the SD rats (Fig. 8A; P < 0.01, SD 24 vs. all other groups). BN 24 rats on the other hand, showed no increase in IR-induced nitrotyrosine formation back to levels observed in sham controls (Fig. 8A). In order to determine whether nitrotyrosine levels were increased in the vulnerable tubular regions of the kidneys, immunohistochemical staining for nitrotyrosine was done in BN sham, SD sham, BN 24, and SD 24 kidneys. This revealed that while there was increased nitrotyrosine immunostaining in the cortico-medullary region of the SD 24 rat kidney, there was no nitrotyrosine present in the BN 24 or respective sham kidneys (Fig. 8B).

(A) Slot-blot analysis for nitrotyrosine showing increased nitrated protein in SD 24 rats compared to sham rats or BN 24 rats. Results are means ± SEM (n = 6 for all groups). ‡ P < 0.01 SD 24 vs. all other groups. (B) Immunohistochemical staining for nitrotyrosine in SD sham, SD 24, BN sham and BN 24 showing increased nitrated proteins in the medulla of kidneys of SD 24 rats compared to BN 24 or sham operated controls
Discussion
In this study, we have shown for the first time that the resistance of the BN rats to renal IR injury is associated with decreased apoptosis, lower O ·−2 levels, decreased lipid peroxidation, and nitrotyrosine formation.
Similar to the original report [25] describing BN’s resistance to IR, we found that compared with SD rats, BN rats had lower plasma creatinine and less evidence of acute tubular necrosis after IR. Our finding that the BN rat is resistant to renal IR injury is consistent with previous reports that have shown a similar pattern for the kidney [25].
In order to determine if the decreased histological damage in the BN rat following IR was associated with decreased programmed cell death, we measured apoptosis by the TUNEL assay in kidney tissue slices. In addition to protection against acute tubular necrosis and preservation of renal function BN rats also displayed a lower level of TUNEL staining, compared to SD rats following IR. This is the first report indicating a differential pattern of apoptosis between BN and SD rats following IR. This indicates that the inherent protection of the BN rats against apoptosis may be partially responsible for improved renal function in renal IR injury.
We were next interested in exploring whether maintenance of pro-oxidant/antioxidant balance contributes to the dramatic resistance of the BN rats to renal IR injury. We thus determined O ·−2 levels by lucigenin-enhanced chemiluminescence in BN and SD sham and IR-operated rats. Although BN shams had slightly higher signals vs. SD sham animals, after IR, there was a significant increase in O ·−2 levels in the SD rats compared to the BN rats. The source of increased superoxide in our model of injury is not exactly clear. There is evidence in the literature that inhibition of NADPH oxidase by apocyanin prevents cerebral [32] and lung [33] IR injury. In addition, inhibition of xanthine oxidase by allopurinol also prevents renal IR injury [34]. Although we have preliminary data indicating that protein levels for the gp91phox subunit of the NADPH oxidase and xanthine oxidase are increased in the SD rats but not the BN rats following IR injury (data not shown), we cannot exclude mitochondrial sources of O ·−2 in our model. While we have not directly measured mitochondrial ROS formation, it is likely that both membrane NADPH oxidase and mitochondrial sources may be responsible for increased O ·−2 levels in the SD rats.
The finding that BN rats had lower O ·−2 levels compared to SD rats following IR injury, correlates well with an earlier study comparing the BN/Mcw rat strain to Dahl S (SS/Mcw) rat strain in a cardiac IR model showing that hearts of Dahl S rats had much higher levels of O ·−2 compared to hearts of BN rats [26]. Recently, using chromosomal substitution studies, Taylor et al. [35] also concluded that the BN-13SS consomic strain is resistant to salt-induced hypertension due to reduced O ·−2 levels compared to the parental Dahl S strain indicating that specific genes in the BN rat are responsible for the decreased reactive oxygen species formation and subsequent protective effect.
The loss of the O ·−2 -scavenging enzymes has been established before in models of renal IR [18, 20] although the effect on the expression of anti-oxidant proteins in BN rats is yet unknown. In our model, in renal tissue of BN rats, there was a preservation of both RNA and protein of the O ·−2 -removal systems (Mn SOD and CuZn SOD) compared to renal tissue of SD rats post IR. Recently, Alfonso et al. [36] have shown that CuZn SOD gene expression can be downreguated by TNF-α via a JNK-/AP1 signaling pathway. Preliminary data from our lab also indicates that TNf-α is preferentially increased in the SD but not in BN rats after renal IR (data not shown). This, in conjunction with our results indicating that RNA levels of CuZn SOD and Mn SOD were preserved preferentially only in the BN rats following IR, suggests that there may be a differential transcriptional regulation of these genes in the two strains. Another possibility is that CuZn SOD and Mn SOD RNA may be degraded more readily in the SD rats compared to the BN rats after IR. Thus, the preservation of the O ·−2 -removal systems in the BN rats may contribute to lower O ·−2 levels in this strain compared to the SD rats.
Oxidation of fatty acids by O ·−2 leads to the formation of the cytotoxic aldehyde, 4-hydroxynonenal (4-HNE) that initiates lipid peroxidation events. In our model, BN rats displayed lower levels of HNE adducts indicating that this strain also had lower lipid peroxidation and overall oxidative stress compared to SD rats after IR. To our knowledge this is the first time that BN rats have shown to have lower lipid peroxide formation in renal IR. There is evidence of increased immunostaining of malondialdehyde (MDA) and 4-HNE, both major aldehydic metabolites of lipid peroxidation within 30 min of warm ischemia in the cortical regions of the kidney [37]. Interestingly, Walker et al. [38] found that while HNE adducts increased after 40 min of bilateral renal ischemia, this index of lipid peroxidation actually decreased after 3 h or 6 h reperfusion. In contrast, in our studies increased HNE adducts were evident even after 24 h of reperfusion in the SD rats. This later increase in HNE adduct formation in our model may be related to increases in both reactive oxygen and reactive nitrogen species because antioxidants and peroxynitrite scavengers have been shown to effectively limit lipid peroxidation in renal IR [39, 40].
The deleterious role of iNOS in acute renal failure has been investigated before using NOS inhibitors such as N (G)-nitro-l-arginine methyl ester (l-NAME) and l-(1-iminoethyl)lysine (l-NIL) [40, 41], antisense oligonucleotides to iNOS [16, 42, 43] or iNOS knockout mice [44, 45] in in vivo and in vitro models of acute renal failure. Previous studies have shown that treatment with the iNOS inhibitor, l-NIL, at a dose that eliminates plasma NO in a mouse model of renal injury lowers serum creatinine levels [12] and that iNOS knockout mice also display a similar phenotype [44]. Our results showing that the BN rat did not show an increase in IR-induced iNOS expression suggests that the lack of induction of this protein is potentially at least partially responsible for the BN’s resistance to IR.
In order to determine whether increased iNOS expression translated to increased NO levels, we measured nitrate + nitrite levels in both plasma and renal tissue homogenates. Surprisingly, in our study plasma NO levels were increased after IR in both the BN and SD strains. The source of the circulating levels of NO in our model is not yet known though it is possible that eNOS-derived NO may partially contribute to the plasma NO. Previous studies have shown that while proximal tubules from iNOS knockout mice are protected against hypoxic insult, tubules from eNOS knockout mice were severely damaged [45], indicating that the source, location, and levels of NO may ultimately be important in determining the level of injury. The constitutively expressed eNOS is produced by endothelial cells in the kidney and eNOS-derived NO plays an important role in modulating vascular tone. Additionally, pretreatment of SD rats with the NO donor molsidomine or supplementation with arginine attenuates renal dysfunction and oxidative stress following IR [46, 47]. In contrast, larger quantities of NO produced by the actions of the iNOS mediates cellular damage and apoptosis in IR injury. In our model, after IR, renal tissue NO levels in the BN rats were similar to BN sham controls while in the SD rats NO metabolites were increased relative to SD sham controls. Thus, the tissue production of NO in our model in the SD rats suggests that excess NO derived from iNOS may ultimately be responsible for the damage in renal IR.
Furthermore, the combination of NO and O ·−2 can be even more detrimental due to the production of the potent tyrosine nitrating and damaging species, peroxynitrite. In our studies, we found a significant increase in nitrotyrosine, a footprint for peroxynitrite formation, in SD rats after renal IR. The BN rats however, had no increase in nitrotyrosine content after IR, suggesting that the BN had overall lower nitrative stress. Additionally, immunohistochemical analysis revealed that the SD rat kidneys had more nitrotyrosine immunostaining compared to BN kidneys following IR injury. This increase was seen not just in the cortico-medullary region (Fig. 8B) but also in the glomeruli (data not shown). This corresponds well with earlier studies showing that nitration in renal IR occurs specifically in proximal and distal tubules [21] and in some cases glomerular tufts and vasculature [48]. At the present time, the critical targets of nitration in our model are not clear. Other investigators have shown that MnSOD is nitrated and inactivated in renal IR injury [21] and that modification of a single tryptophan residue by peroxynitrite–carbon dioxide or myeloperoxidase–hydrogen peroxide–nitrite can nitrate/oxidize CuZn SOD [49, 50]. The specific loss of these proteins in the SD rat compared to the BN rat after IR in our study suggest that it is possible that in addition to the different transcriptional regulation, these proteins could also be differentially targeted for oxidative and nitrative modifications in the BN and SD rats in IR injury.
In summary, in this study, we have shown for the first time that the resistance of the BN rat to renal IR is associated with the lack of detectable oxidant status and preserved antioxidant defenses. These findings provide a possible basis for understanding the genetic control of resistance to renal IR injury and may be useful in designing clinical strategies that target oxidant systems or utilize antioxidant systems to limit acute renal failure.
References
Edelstein CL, Ling H, Schrier RW (1997) The nature of renal cell injury. Kidney Int 51:1341–1351
Lameire N (2005) The pathophysiology of acute renal failure. Crit Care Clin 21:197–210
Lameire N, Van Biesen W, Vanholder R (2005) Acute renal failure. Lancet 365:417–430
Chien CT, Lee PH, Chen CF et al (2001) De novo demonstration and co-localization of free-radical production and apoptosis formation in rat kidney subjected to ischemia/reperfusion. J Am Soc Nephrol 12:973–982
McCord JM, Roy RS (1982) The pathophysiology of superoxide: roles in inflammation and ischemia. Can J Physiol Pharmacol 60:1346–1352
McCord JM, Roy RS, Schaffer SW (1985) Free radicals and myocardial ischemia. The role of xanthine oxidase. Adv Myocardiol 5:183–189
Unal D, Yeni E, Erel O et al (2002) Antioxidative effects of exogenous nitric oxide versus antioxidant vitamins on renal ischemia reperfusion injury. Urol Res 30:190–194
Wink DA, Hanbauer I, Krishna MC et al (1993) Nitric oxide protects against cellular damage and cytotoxicity from reactive oxygen species. Proc Natl Acad Sci USA 90:9813–9817
Wink DA, Miranda KM, Espey MG et al (2001) Mechanisms of the antioxidant effects of nitric oxide. Antioxid Redox Signal 3:203–213
Martinez-Mier G, Toledo-Pereyra LH, Bussell S et al (2000) Nitric oxide diminishes apoptosis and p53 gene expression after renal ischemia and reperfusion injury. Transplantation 70:1431–1437
Chatterjee PK, Patel NS, Kvale EO et al (2002) Inhibition of inducible nitric oxide synthase reduces renal ischemia/reperfusion injury. Kidney Int 61:862–871
Zahmatkesh M, Kadkhodaee M, Arab HA et al (2006) Effects of co-administration of an iNOS inhibitor with a broad-spectrum reactive species scavenger in rat renal ischemia/reperfusion injury. Nephron Exp Nephrol 103:e119–e125
Beckman JS (2002) Protein tyrosine nitration and peroxynitrite. FASEB J 16:1144
Beckman JS, Chen J, Ischiropoulos H et al (1994) Oxidative chemistry of peroxynitrite. Methods Enzymol 233:229–240
Beckman JS, Ischiropoulos H, Zhu L et al (1992) Kinetics of superoxide dismutase- and iron-catalyzed nitration of phenolics by peroxynitrite. Arch Biochem Biophys 298:438–445
Goligorsky MS, Brodsky SV, Noiri E (2002) Nitric oxide in acute renal failure: NOS versus NOS. Kidney Int 61:855–861
Gow AJ, Ischiropoulos H (2001) Nitric oxide chemistry and cellular signaling. J Cell Physiol 187:277–282
Davies SJ, Reichardt-Pascal SY, Vaughan D et al (1995) Differential effect of ischaemia-reperfusion injury on anti-oxidant enzyme activity in the rat kidney. Exp Nephrol 3:348–354
Jassem W, Fuggle SV, Rela M et al (2002) The role of mitochondria in ischemia/reperfusion injury. Transplantation 73:493–499
Dobashi K, Ghosh B, Orak JK et al (2000) Kidney ischemia-reperfusion: modulation of antioxidant defenses. Mol Cell Biochem 205:1–11
Cruthirds DL, Novak L, Akhi KM et al (2003) Mitochondrial targets of oxidative stress during renal ischemia/reperfusion. Arch Biochem Biophys 412:27–33
Cruthirds DL, Saba H, MacMillan-Crow LA (2005) Overexpression of manganese superoxide dismutase protects against ATP depletion-mediated cell death of proximal tubule cells. Arch Biochem Biophys 437:96–105
Yin M, Wheeler MD, Connor HD et al (2001) Cu/Zn-superoxide dismutase gene attenuates ischemia-reperfusion injury in the rat kidney. J Am Soc Nephrol 12:2691–2700
Baker JE, Konorev EA, Gross GJ et al (2000) Resistance to myocardial ischemia in five rat strains: is there a genetic component of cardioprotection?. Am J Physiol Heart Circ Physiol 278:H1395–1400
Basile DP, Donohoe D, Cao X et al (2004) Resistance to ischemic acute renal failure in the Brown Norway rat: a new model to study cytoprotection. Kidney Int 65:2201–2211
Shi Y, Hutchins W, Ogawa H et al (2005) Increased resistance to myocardial ischemia in the Brown Norway vs. Dahl S rat: role of nitric oxide synthase and Hsp90. J Mol Cell Cardiol 38:625–635
Blydt-Hansen TD, Katori M, Lassman C et al (2003) Gene transfer-induced local heme oxygenase-1 overexpression protects rat kidney transplants from ischemia/reperfusion injury. J Am Soc Nephrol 14:745–754
Pieper GM, Nilakantan V, Zhou X et al (2005) Treatment with {alpha}-phenyl-N-tert-butylnitrone, a free radical-trapping agent, abrogates inflammatory cytokine gene expression during alloimmune activation in rat cardiac allografts. J Pharmacol Exp Ther 312:774–779
Khadour FH, Panas D, Ferdinandy P (2002) Enhanced NO and superoxide generation in dysfunctional hearts from endotoxemic rats. Am J Physiol Heart Circ Physiol 283:H1108–H1115
Nilakantan V, Zhou X, Hilton G et al (2005) Hierarchical change in antioxidant enzyme gene expression and activity in acute cardiac rejection: role of inducible nitric oxide synthase. Mol Cell Biochem 270:39–47
Nilakantan V, Halligan NL, Nguyen TK et al (2005) Post-translational modification of manganese superoxide dismutase in acutely rejecting cardiac transplants: role of inducible nitric oxide synthase. J Heart Lung Transplant 24:1591–1599
Wang Q, Tompkins KD, Simonyi A et al (2006) Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res 1090:182–189
Dodd OJ, Pearse DB (2000) Effect of the NADPH oxidase inhibitor apocynin on ischemia-reperfusion lung injury. Am J Physiol Heart Circ Physiol 279:H303–H312
Rhoden E, Teloken C, Lucas M et al (2000) Protective effect of allopurinol in the renal ischemia-reperfusion in uninephrectomized rats. Gen Pharmacol 35:189–193
Taylor NE, Glocka P, Liang M et al (2006) NADPH oxidase in the renal medulla causes oxidative stress and contributes to salt-sensitive hypertension in Dahl S rats. Hypertension 47:692–698
Afonso V, Santos G, Collin P et al (2006) Tumor necrosis factor-alpha down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free Radic Biol Med 41:709–721
Eschwege P, Paradis V, Conti M et al (1999) In situ detection of lipid peroxidation by-products as markers of renal ischemia injuries in rat kidneys. J Urol 162:553–557
Walker LM, York JL, Imam SZ et al (2001) Oxidative stress and reactive nitrogen species generation during renal ischemia. Toxicol Sci 63:143–148
Erdogan H, Fadillioglu E, Yagmurca M et al (2006) Protein oxidation and lipid peroxidation after renal ischemia-reperfusion injury: protective effects of erdosteine and N-acetylcysteine. Urol Res 34:41–46
Noiri E, Nakao A, Uchida K et al (2001) Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol 281:F948–F957
Yu L, Gengaro PE, Niederberger M et al (1994) Nitric oxide: a mediator in rat tubular hypoxia/reoxygenation injury. Proc Natl Acad Sci USA 91:1691–1695
Noiri E, Peresleni T, Miller F et al (1996) In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J Clin Invest 97:2377–2383
Peresleni T, Noiri E, Bahou WF et al (1996) Antisense oligodeoxynucleotides to inducible NO synthase rescue epithelial cells from oxidative stress injury. Am J Physiol Renal Physiol 270:F971–F977
Ling H, Edelstein C, Gengaro P et al (1999) Attenuation of renal ischemia-reperfusion injury in inducible nitric oxide synthase knockout mice. Am J Physiol Renal Physiol 277:F383–F390
Ling H, Gengaro PE, Edelstein CL (1998) Effect of hypoxia on proximal tubules isolated from nitric oxide synthase knockout mice. Kidney Int 53:1642–1646
Chander V, Chopra K (2006) Possible role of nitric oxide in the protective effect of resveratrol in 5/6th nephrectomized rats. J Surg Res 133:129–135
Chander V, Chopra K (2006) Renal protective effect of molsidomine and L-arginine in ischemia-reperfusion induced injury in rats. J Surg Res 128:132–139
Mark LA, Robinson AV, Schulak JA (2005) Inhibition of nitric oxide synthase reduces renal ischemia/reperfusion injury. J Surg Res 129:236–241
Yamakura F, Matsumoto T, Fujimura T et al (2001) Modification of a single tryptophan residue in human Cu,Zn-superoxide dismutase by peroxynitrite in the presence of bicarbonate. Biochim Biophys Acta 1548:38–46
Yamakura F, Matsumoto T, Ikeda K et al (2005) Nitrated and oxidized products of a single tryptophan residue in human Cu,Zn-superoxide dismutase treated with either peroxynitrite-carbon dioxide or myeloperoxidase-hydrogen peroxide-nitrite. J Biochem (Tokyo) 138:57–69
Author information
Authors and Affiliations
Corresponding author
Additional information
This work was supported in part by a Medical College of Wisconsin-Research Affairs Committee Grant to V. Nilakantan, and by divisional funds to V. Nilakantan and B.D. Shames.
Rights and permissions
About this article
Cite this article
Nilakantan, V., Hilton, G., Maenpaa, C. et al. Favorable balance of anti-oxidant/pro-oxidant systems and ablated oxidative stress in Brown Norway rats in renal ischemia-reperfusion injury. Mol Cell Biochem 304, 1–11 (2007). https://doi.org/10.1007/s11010-007-9480-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-007-9480-z