
Abstract
Thyroxine can cause cardiac hypertrophy by activating growth factors, such as IGF-I (insulin-like growth factor-I). Since oxidative stress is enhanced in the hyperthyroidism, it would control protein expression involved in this hypertrophy. Male Wistar rats were divided into four groups: (I) control, (II) vitamin E-supplemented (20 mg/kg/day subcutaneous), (III) hyperthyroid (thyroxine 12 mg/l, in drinking water), and (IV) hyperthyroid + vitamin E. After 4 weeks, the contractility and relaxation indexes of left ventricle (LV), and cardiac mass were increased by 54%, 60%, and 60%, respectively, in hyperthyroid group. An increase in lipid peroxidation (around 40%), and a decrease in total glutathione (by 20%) was induced by thyroxine and avoided by vitamin E administration. Superoxide dismutase (SOD) and glutathione-S-transferase (GST) activities were increased (by 83% and 54%, respectively) in hyperthyroid, and vitamin E avoided changes in SOD. Protein expression of SOD, GST, and IGF-I receptor (IGF-IR) were increased (by 87%, 84%, and 60%, respectively) by thyroxine, and vitamin E promoted a significant reduction in SOD and IGF-IR expression (by 36% and 17%, respectively). These results indicate that oxidative stress is involved in cardiac hypertrophy, and suggest a role for IGF-IR as a mediator of this adaptive response in experimental hyperthyroidism.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Thyrotoxic cardiomyopathy is associated with the effects of thyroid hormones in cardiovascular system [1]. The thyroid hormones may cause hemodynamic changes, including a decrease in systemic vascular resistance, an increase in myocardial contractility, an elevation in cardiac output, a widening of pulse pressure, cardiac arrhythmias, and atrial fibrillation [2]. These effects are due to thyroid hormone-mediated actions on cardiac myocyte gene expression, as well as direct non-genomic effects of thyroid hormones on the cells [2].
Is well recognized a hyperkinetic cardiocirculatory state in hyperthyroidism, and this situation may increase oxygen demand to myocardium, resulting in the increased production of reactive oxygen species (ROS) [1]. ROS can play an important role in mediating the effects of thyroid hormones, especially those related to electrical and mechanical activity of the heart [2]. In order to counteract ROS production, the enzymatic and nonenzymatic antioxidant defense systems are present to minimize the oxidative damage due to these species [3, 4]. However, when ROS generation is exacerbated and overcome the antioxidant capacity of cells, oxidative stress develops, leading to potential tissue damage [5]. Evidence from experimental, as well as clinical studies, point out the involvement of oxidative stress in the pathogenesis of cardiac dysfunction, such as cardiac hypertrophy and failure [3]. There is a positive correlation between hyperthyroidism and cardiac hypertrophy [6], and ROS may be involved in this adaptive answer [7]. However, the molecular mechanisms of ROS mediating these heart changes are still poorly understood.
In a previous study, we have demonstrated a positive correlation among oxidative stress, cardiac hypertrophy, and left ventricular diastolic dysfunction in experimental hyperthyroidism, indicating that ROS may contribute to the progression to heart failure in this model [8]. In hyperthyroidism, however, there is scarce information about the induction of gene expression of redox-sensitive factors in the hypertrophic heart, including insulin-like growth factor-I (IGF-I).
IGF-I may be synthesized by organs, including heart and vessels, and can act as an autocrine and paracrine factor. IGF-I plays an essential role in the regulation of cellular growth and development [9]. Its interactions with the IGF-I receptors (IGF-IR), which are abundantly expressed in myocardium, stimulates both proliferation and differentiation of cardiomyocytes in vivo and in vitro [10], and this could contribute to the hypertrophic responses in myocardium [11, 12]. There is also experimental evidence for the role of IGF-I in the increase in calcium sensitivity of cardiac myofilaments and prevention of apoptosis [13]. In vascular muscle cell, IGF-IR transcription angiotensin II-stimulated was demonstrated to be completely abolished by antioxidants, suggesting involvement of redox signaling [14]
In the present study, we hypothesized that the use of a classical antioxidant (vitamin E) decreases oxidative stress, reducing IGF-IR expression in myocardium, avoiding the development of myocardial hypertrophy and failure in experimental hyperthyroidism.
Material and methods
Animals
Forty male Wistar rats (200 ± 20 g) were obtained from the Central Animal House of the Universidade Federal do Rio Grande do Sul, Brazil. Animals were housed in plastic cages (five animals each) and received water and pelleted food ad libitum. They were maintained under standard laboratory conditions (controlled temperature of 21°C, 12 h light/dark cycle). Animals were weekly weighed to follow body weight gain, during the experimental protocol (28 days). They were divided into four groups (n = 10/group): (I) control, that received water ad libitum and subcutaneous injections of mineral oil; (II) vitamin E-supplemented, that received subcutaneous injections of vitamin E (20 mg/Kg/day, in mineral oil) [15]; (III) hyperthyroid, which received l-thyroxine (T4) (12 mg/l in drinking water) [16]; (IV) hyperthyroid + vitamin E, which received T4 and vitamin E, at the same conditions as above described.
Hemodynamic measurements and cardiac hypertrophy development
Cardiac hemodynamics was assessed at the end of the 4th week of treatment. In brief, rats were anesthetized (ketamine 90 mg/kg; xylazine 10 mg/kg, i.p.) and the right carotid artery was cannulated with a PE 50 catheter connected to a strain gauge transducer (Narco Biosystem Pulse Transducer RP-155, Houston, Texas, USA) linked to a pressure amplifier (HP 8805C, Hewlett Packard, USA). Pressure readings were taken in a microcomputer equipped with an analogue-to-digital conversion board (CODAS 1 kHz sampling frequency, Dataq Instruments, Inc., Akron, Ohio, USA). The catheter was advanced into left ventricle (LV) for recording the left ventricular systolic pressure (LVSP, mmHg), the left ventricular end diastolic pressure (LVEDP, mmHg), and the positive and negative pressure derivatives (±dP/dt). Positive derivative of ventricular pressure is considered as the contractility index and the negative derivative is the relaxation index. The cardiac hypertrophy was evaluated by heart weight (in mg) to body weight (in g) ratio.
Tissue preparation
Four weeks after treatment, rats were decapitated and the hearts were rapidly excised, weighed, and homogenized (1.15% w/v KCl and phenyl methyl sulphonyl fluoride PMSF 20 mmol/l) in Ultra-Turrax. The suspension was centrifuged at 600 g for 10 min at 0−4°C to remove the nuclei and cell debris [17] and supernatants were used for the assay of lipid peroxidation and enzymatic activity. In the moment of the sacrifice, cardiac tissue samples were also removed and frozen at −80°C for the evaluation of glutathione content and protein expression. Lung and liver were also taken and weighed in order to estimate the congestion in these organs in terms of wet weight of the organ to body weight ratio.
Thyroxine concentration
Blood samples were collected by cardiac puncture, and immediately centrifuged at 1,000 g for 10 min. Serum thyroxine concentration was estimated by chemiluminescence using the Immunolite 2,000 kit (Biomedical Technologies, Inc., Strougerton, MA, USA) at Weinmann Clinical Analysis Laboratory.
Tert-butyl hydroperoxide-initiated chemiluminescence
Chemiluminescence (CL) was measured in a liquid scintillation counter in the out-of-coincidence mode (LKB Rack Beta Liquid Scintillation Spectrometer 1215, LKB—Produkter AB, Sweden). Homogenates were placed in low-potassium vials at a protein concentration of 0.5–1.0 mg/ml in a reaction medium consisting of 120 mmol/l KCl, 30 mmol/l phosphate buffer (pH = 7.4). Measurements were started by the addition of 3 mmol/l tert-butyl hydroperoxide and data expressed as counts per second per milligram of protein (cps/mg protein) [18].
Thiobarbituric acid reactive substances method (TBARS)
For the TBARS assay, trichloroacetic acid (10% w/v) was added to the homogenate to precipitate proteins and to acidify samples [19]. This mixture was then centrifuged (1,000 g, 3 min). The protein-free sample was extracted and thiobarbituric acid (0.67% w/v) was added to the reaction medium. Tubes were placed in a water bath (100°C) for 15 min. Absorbance was read at 535 nm in a spectrophotometer. Commercially available malondialdehyde was used as a standard. Results were expressed as micromoles per milligram of protein.
Determination of total glutathione content
To determine total glutathione content, tissue was deproteinized with 2 mol/l perchloric acid, centrifuged for 10 min at 1,000 g, and the supernatant was neutralized with 2 mol/l potassium hydroxide. The reaction medium contained 100 mmol/l phosphate buffer (pH 7.2), 2 mmol/l nicotinamide dinucleotide phosphate acid, 0.2 U/ml glutathione reductase, 70 μmol/l 5,5′ dithiobis (2-nitro benzoic acid), and was measured at 420 nm. The results were expressed in nmol per milligram of protein [20].
Determination of antioxidant enzyme activities
Superoxide dismutase (SOD) activity, expressed as units per milligram of protein, was based on the inhibition of superoxide radical reaction with pyrogallol [21]. Catalase (CAT) activity was determined by following the decrease in hydrogen peroxide (H2O2) absorbance at 240 nm. It was expressed as nanomol of H2O2 reduced per minute per milligram of protein [22]. Glutathione-S-transferase (GST) activity, expressed as nanomol per milligram of protein, was measured by the rate of formation of dinitrophenyl-S-glutathione at 340 nm [23].
Western blot analysis
Tissue homogenization, electrophoresis, and protein transference were performed as described elsewhere [8, 24] The membranes were processed for immunodetection using rabbit anti-CAT polyclonal antibody, sheep anti-Cu/Zn SOD polyclonal antibody, rabbit anti-GST polyclonal antibody, and rabbit anti-IGF-IR polyclonal antibody as primary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). The bound primary antibodies were detected using rabbit anti-sheep or goat anti-rabbit horseradish peroxidase-conjugate secondary antibodies and membranes were revealed for chemiluminescence. The autorradiographs generated were quantitatively analyzed with an image densitometer (Imagemaster VDS CI, Amersham Biosciences Europe, IT). The molecular weights of the bands were determined by reference to a standard molecular weight marker (RPN 800 rainbow full range Bio-Rad, CA, USA). The results from each membrane were normalized through Ponceau red method [25].
Determination of protein concentration
Protein was measured by the method of Lowry [26], using bovine serum albumin as standard.
Statistical analysis
Data were expressed as mean ± SD. To compare multiple groups, we used one way ANOVA with post-hoc Student-Newmann-Keuls. The correlation between two variables was analyzed by Pearson’s correlation. Values of P < 0.05 were considered statistically significant.
Results
Hyperthyroidism and cardiac remodeling
At the end of 4th week post-treatment period, serum thyroxine levels (in ng/ml) were significantly higher in hyperthyroid (24.0 ± 0.7) and hyperthyroid + vitamin E (19.8 ± 0.8) than control (3.8 ± 0.1) and vitamin E-supplemented (3.9 ± 0.2). However, no statistical difference was found between the two hyperthyroid groups (III and IV). In hyperthyroid, animal’s body weight was decreased by 13% in comparison to control group, and vitamin E administration avoided this change ( Table 1). Thyroxine has induced cardiac hypertrophy as detected by heart to body weight ratio (mg/g), which was 60% higher in hyperthyroid group than control. However, after vitamin E treatment, heart to body weight ratio in group IV has shown a 23% reduction as compared to hyperthyroid, and it was not different from the control (P < 0.05) (Table 1). In terms of intraventricular pressure, hyperthyroidism induced elevation of left ventricular systolic pressure (LVSP) and left ventricular end diastolic pressure (LVEDP) by 44% and 128%, respectively, as compared to control. LVSP was reduced by 20% in group IV, but no significant decrease in LVEDP was observed in this group (Table 1). The positive derivative (+dP/dt) increased by 54%, and negative derivative (−dP/dt) by 60% in the hyperthyroid. However, vitamin E administration, in the hyperthyroid animals, decreased +dP/dt by 23%, as well as −dP/dt by 28% (Table 1).
Oxidative damage to lipids
Changes in lipid peroxidation markers are summarized in Fig. 1. Hyperthyroid state caused a significant elevation in myocardial lipid peroxidation products, as indicated by chemiluminescence (32%), and TBARS (47%), as compared to control (P < 0.05) (Fig. 1A and B). However, in group IV, CL and TBARS were reduced by 25% and 33%, respectively, as compared to group III (P < 0.05). It was found that hypertrophy index showed a significant positive correlation with CL (r = 0.73, P < 0.05) and TBARS values (r = 0.8, P < 0.05).

Myocardial markers of oxidative damage to membrane lipids: chemiluminescence (A), and TBARS (B). Data as mean ± SD from 10 animals in each group. * significantly different from I (P < 0.05). • significantly different from III (P < 0.05). # significantly different from II (P < 0.05). I = Control; II = vitamin supplemented E; III = hyperthyroid; IV = hyperthyroid + vitamin E
Total glutathione content
There was a 20% decrease in the total GSH, in hyperthyroid as compared to control (P < 0.05) (Table 2). Vitamin E reversed these effects. Vitamin E administration increased total GSH content in euthyroid animals (P < 0.05) (Table 2).
Antioxidant enzyme activities and protein expression
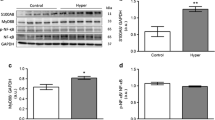
The l-thyroxine treatment to rats resulted in no significant change in CAT activity (Table 2) and protein expression (Fig. 2A) among the experimental groups. There was elevation in SOD activity in the hyperthyroid group, which was 83% higher than control (Table 2). The protein levels, evaluated by Western blot, of Cu/Zn SOD were elevated by 87% in hyperthyroid group as compared to control (P < 0.05) (Fig. 2B). The treatment of hyperthyroid animals with vitamin E reduced SOD activity by 36% (Table 2), and protein expression decreased by 16% in comparison to hyperthyroid (Fig. 2B). Hyperthyroidism has also induced an increase in GST activity (Table 2) and protein expression (Fig. 2C) (by 54% and 84%, respectively) as compared to control (P < 0.05). Group IV showed augmented GST activity as compared to group II (82%) (Table 2, Fig. 2C). No difference was found among groups II, III, and IV in terms of GST protein levels (Fig. 2C). In the group IV, GST activity was additionally increased in comparison to group II (Table 2).

Western blot analysis in cardiac homogenates using CAT antibody (A), Cu/Zn SOD antibody (B), GST antibody (C), and IGF-IR antibody (D). Data as mean ± SD from 10 animals in each group (one representative gel of five Western blot experiments, showing two bands for each experimental group). * significantly different from I (P < 0.05). • significantly different from III (P < 0.05). I = Control; II = vitamin E supplemented; III = hyperthyroid; IV = hyperthyroid + vitamin E
IGF-IR protein expression
IGF-IR myocardial protein concentration analyzed by Western blot was higher (60%) in hyperthyroid group than control. Vitamin E treatment decreased (by 17%) IGF-IR protein expression induced by l-thyroxine (Fig. 2D). IGF-IR protein levels were positively correlated with the hypertrophy index and LVEDP values (r = 0.90, P < 0.05; r = 0.74, P < 0.05, respectively).
Discussion
The presence of cardiovascular alterations and ventricular remodeling phenomenon in clinical and experimental hyperthyroidism is well documented in literature [27]. We have demonstrated, in a previous work, that ventricular dysfunction in experimental hyperthyroidism is correlated to oxidative damage and antioxidant enzyme changes in protein expression [8]. Thus, we have suggested that oxidative stress is involved in these ventricular alterations. Therefore, treatment with antioxidants would reduce oxidative damage, and thus, improve cardiac function. The present study was designed to test this hypothesis by administration of vitamin E (a classical antioxidant) and to explore the possible physiological mechanism.
The morphometric and functional changes in this study were typical of hyperthyroidism, including decreased body weight, cardiac hypertrophy, and increased ±dP/dt. Collectively, these data confirmed that chronic hyperthyroidism was achieved. Cardiac hypertrophy in hyperthyroidism is associated with an altered pattern of gene expression of many proteins, such as myosin heavy chain, calcium transport/regulation proteins, and many other components of excitation-contraction coupling [28]. Angiotensin II, that induces ROS production, has been also involved in cardiac hypertrophy in hyperthyroidism [29]. LVSP elevation, observed in this study in hyperthyroid rats, may be ascribed not only to the increased heart to body weight ratio, but also to enhanced the contractility index (+dP/dt). These findings corroborate with data available in literature in experimental hyperthyroidism [30], as well as in hyperthyroid patients [31]. However, in human overt hyperthyroidism, myocardial contractility does not play a major role in increasing LV performance, which is instead predominantly sustained by increased preload with enhanced LV diastolic function [31]. Increased diastolic relaxation, contractility, and heart rate cause an elevation in cardiac output. Our data also demonstrated an increased relaxation index (−dP/dt) in the hyperthyroid rats. Besides that, LVEDP is greatly enhanced in these animals, probably reflecting an augmented residual volume. Thyroxine treatment leads to liver and lung congestion, indicating that these animals were in a congestive heart failure stage. In a previous study, we monitored LVEDP from the first to the 4th week of T4 administration, and it was increased significantly only in the last week of treatment [8]. These data suggest that besides of the amelioration in ventricular performance (±dP/dt), at this stage, these cardiac changes were not enough to compensate the increased preload experimented by these hyperthyroid hearts and congestive heart failure which appeared [8]. This alteration coincided with increased myocardial oxidative stress. In fact, morphometric and hemodynamic parameters were improved when vitamin E was administered, especially hypertrophy and organ congestion index. LVEDP did not show improvement, however, the ±dP/dt, that is a more sensitive measurement, returned to normal levels with vitamin E treatment.
Major outcome of the present study was that myocardial oxidative stress was increased, concomitant with the development of cardiac hypertrophy and failure, and the treatment with a classical antioxidant, vitamin E, reduced oxidative stress and ventricular dysfunction. Our results show significant increase in myocardial lipid peroxidation (LPO) in hyperthyroid group by means of two different methods. It was also shown a positive correlation between oxidative damage and cardiac hypertrophy, confirmed by the use vitamin E that was effective in reducing cardiac mass.
In terms of antioxidant enzymes, no changes were found in CAT activity and concentration in the myocardium of hyperthyroid rats. Myocardial SOD activity and expression were found to be increased in the hyperthyroid animals as compared to controls. However, SOD activity and expression were decreased with vitamin E treatment. These results suggest that superoxide radical’s concentration is increased in hyperthyroid, and this ROS probably modulates the regulation of this enzyme [32]. The augmented activity and protein expression of GST found in hyperthyroid and vitamin E groups can reflect the important role of this enzyme in the detoxification of xenobiotics, and therefore, could be involved in l-thyroxine and vitamin E metabolism. GST activity involves an intense participation of GSH in blocking the peroxidation process, altering cellular redox satus. GSH depletion would lead to intracellular signaling for the protein expression regulation [32]. A total GSH decrease was noticed in this study, and it may reinforce that oxidative stress is taking place in cardiac tissue of hyperthyroid rats [33]. We found a negative correlation between total GSH and cardiac mass (r = −0.85, P < 0.05), suggesting that cardiac hypertrophy development is associated with GSH depletion.
Hyperthyroidism can cause increase in cardiac mass by activating growth factors, such as insulin-like growth factor I (IGF-I) [11]. Our results have shown an increase in IGF-IR protein expression in group III. However, vitamin E administration reduced IGF-IR levels in group IV as compared to group III. Since vitamin E is considered as a classical antioxidant, this result suggests that oxidative stress participates in the control of IGF-IR expression in hyperthyroidism. We have also demonstrated a negative correlation between total GSH content and IGF-IR protein expression (r = −0.55, P < 0.05). The data suggest that IGF-IR redox-dependent protein expression may be through an unbalance in GSH metabolism. GSH depletion could act as a triggering event for cardiac mass increment, since IGF-IR protein level was strongly correlated with the hypertrophy index values (r = 0.90, P < 0.05). These data indicate a role for oxidative stress in IGF-IR protein expression and myocardial hypertrophic answer in experimental hyperthyroidism.
In summary, chronic treatment with thyroxine induces myocardial hypertrophy and failure by oxidative stress induction. Thyroxine may activate ROS-sensitive pathways that culminate in the expression of some mediator factors for the myocardial hypertrophy, such as IGF-IR. Vitamin E administration ameliorates the morphometric and hemodynamic impairment induced by thyroxine, exhibiting a beneficial role in the ventricular dysfunction. The overall results point out to a perspective of therapeutical strategy targeting oxidative stress reduction, to decrease its impact in terms of the progression from cardiac hypertrophy to failure in hyperthyroid patients.
References
Venditti P, Di Meo S (2006) Thyroid hormones-induced oxidative stress. Cell Mol Life Sci 63:414–434
Klein I, Ojamaa K (2001) Thyroid hormone and cardiovascular system. N Engl J Med 344:501–509
Li T, Danelisen I, Belló-Klein A, Singal PK (2000) Effects of probucol on changes of antioxidant enzymes in adriamycin induced cardiomyopathy in rats. Cardiovasc Res 46:523–530
Das K, Chainy GB (2001) Modulation of rat liver mitochondrial antioxidant defence system by thyroid hormone. Biochim Biophys Acta 1537:1–13
Esterbauer H, Shaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde, and related aldehydes. Free Rad Biol Med 11:81–128
Donatelli M, Assennato P, Abbadi V, Bucalo ML, Compagno V, Lo Vecchio S, Messina L, Russo V, Schembri A, Torregrossa V, Licata G (2003) Cardiac changes in subclinical and overt hyperthyroid women: retrospective study. Int J Cardiol 90:159–164
Sawyer DB, Siwik DA, Xiao L, Pimentel DR, Singh K, Colucci WS (2002) Role of oxidative stress in myocardial hypertrophy and failure. J Mol Cell Cardiol 34:379–389
Araujo ASR, Ribeiro MFM, Enzveiler A, Schenkel P, Fernandes TRG, Partata WA, Irigoyen MC, Llesuy S, Belló-Klein A (2006) Myocardial antioxidant enzyme activities and concentration and glutathione metabolism in experimental hyperthyroidism. Mol Cell Endocrinol 249:133–139
McMullen JR, Shioi T, Huang W (2004) The insulin-like growth factor 1 receptor induces physiological heart growth via phosphoinositide 3-kinase (p110α) pathway. J Biol Chem 6:4782–4793
Das DK, Maulik N, Engelman RM (2004) Redox regulation of angiotensin II signaling in the heart. J Cell Med 8:144–152
Kuo WW, Chu CY, Wu CH, Lin JA, Liu JY, Hsieh YH, Ueng KC, Lee SD, Hsieh DJ, Hsu HH, Chen LM, Huang CY (2005) Impaired IGF-I signaling of hypertrophic hearts in the developmental phase of hypertension in genetically hypertensive rats. Cell Biochem Funct 23:325–331
Dorn II GW, Force T (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115:527–537
Delafontaine P, Brink M (2000) The growth hormone and insulin-like growth factor 1 axis in heart failure. Ann Endocrinol (Paris) 61:22–26
Du J, Peng T, Scheidegger KJ, Delafontaine P (1999) Angiotensin II activation of insulin-like growth factor-1 receptor transcription is mediated by a tyrosine kinasedependent redox-sensitive mechanism. Arterioscler Thromb Vasc Biol 19:2119–2126
Chitra KC, Mathur PP (2004) Vitamin E prevents nonylphenol-induced oxidative stress in testis of rats. Indian J Exp Biol 42:220–223
Ladenson PW, Kieffer JD, Farwell AP, Ridgway EC (1986) Modulation of myocardial L-triiodothyronine receptors in normal, hypothyroid and hyperthyroid rats. Metabolism 35:5–12
Llesuy SF, Milei J, Molina H, Boveris A, Milei S (1985) Comparison of lipid peroxidation and myocardial damage induced by adriamycin and 4′-epiadrimycin in mice. Tumori 71:241–249
Gonzalez-Flecha B, Llesuy S, Boveris A (1991) Hydroperoxide-initiated chemiluminescence: an assay for oxidative stress in biopsies of liver, heart and muscle. Free Rad Biol Med 10:41–47
Buege JA, Aust SD (1978) Microsomal lipid peroxidation. Methods Enzymol 52:302–309
Akerboom T, Sies H (1981) Assay of glutathione disulfide and glutathione mixed disulfides in biological samples. Methods Enzymol 77:373–382
Marklund S (1985) Pyrogallol autooxidation. In: Greenwald RA (ed) Handbook of methods for oxygen radical research. CRC Press, Boca Raton, FL, pp 243–247
Aebi H (1984) Catalase in vitro. Methods Enzymol 105:121–126
Mannervik B, Gluthemberg C (1981) Glutathione transferase. Methods Enzymol 77:231–235
Laemmli V (1970) Cleavage of structural proteins during the assembly of the head of bacteriophageT4. Nature 227:680–685
Klein D, Kern RM, Sokol RZ (1995) A method for quantification and correction of proteins after transfer to immobilization membranes. Biochem Mol Biol 36:1
Lowry OH, Rosebrough AL, Farr AL, Randall R (1951) Protein measurement with the folin phenol reagent. J Biol Chem 193:265–275
Dörr M, Wolff B, Robinson DM, John U, Lüdemann J, Meng M, Felix SB, Völzke H (2005) The association of thyroid function with cardiac mass and left ventricular hypertrophy. J Clin Endocrinol. Metabolism 90:673–677
Kiss E, Jakab G, Kranias EG, Edes I (1994) Thyroid hormone-induced alternations in phospholamban protein expression: regulatory effects on sarcoplasmic reticulum Ca+2 transport and myocardial relaxation. Circ Res 75:245–251
Hu LW, Benvenuti LA, Liberti EA, Carneiro-Ramos MS, Barreto-Chaves ML (2003) Thyroxine-induced cardiac hyperthrophy: influence of adrenergic nervous system versus renin- angiotensin system on myocyte remodeling. Am J Physiol Regul Integ. Comp Physiol 285:1473–1480
Kuzman AJ, Vogelsang KA, Thomas AT, Martin Gerdes A (2005) L-Thyroxine activates Akt signaling in the heart. J Mol Cell Cardiol 39:251–258
Palmieri EA, Fazio S, Palmieri V, Lombardi G, Biondi B (2004) Myocardial contractility and total arterial stiffness in β1-adrenergic blockade. Europ J End 150:757–762
Das K, Chainy GB (2004) Thyroid hormone influences antioxidant defense system in adult rat brain. Neurochem Rese 29:1755–1766
Dincer Y, Akca T, Alademir Z, Ilkova H (2002) Effect of oxidative stress on glutathione pathway in red blood cells from patients with insulin dependent diabetes mellitus. Metabolism 51:1360–1362
Acknowledgments
This work was supported by CNPq and FAPERGS, Brazilian Research Agencies. We would like to acknowledge Weinmann Clinical Analysis Laboratory for thyroid hormone measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Araujo, A.S.R., Enzveiler, A.T., Schenkel, P. et al. Oxidative stress activates insulin-like growth factor I receptor protein expression, mediating cardiac hypertrophy induced by thyroxine. Mol Cell Biochem 303, 89–95 (2007). https://doi.org/10.1007/s11010-007-9459-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-007-9459-9