
Abstract
Hyperthyroidism can lead to the activation of proteins which are associated with inflammation, apoptosis, hypertrophy, and heart failure. This study aimed to explore the inflammatory and apoptotic proteins involved in the hyperthyroidism-induced cardiac hypertrophy establishment. Male Wistar rats were divided into control and hyperthyroid (12 mg/L L-thyroxine, in drinking water for 28 days) groups. The expression of inflammatory and apoptotic signaling proteins was quantified in the left ventricle by Western blot. Hyperthyroidism was confirmed by evaluation of T3 and T4 levels, as well as cardiac hypertrophy development. There was no change in the expression of HSP70, HIF1-α, TNF-α, MyD88, p-NFκB, NFκB, p-p38, and p38. Reduced expression of p53 and PGC1-α was associated with increased TLR4 and decreased IL-10 expression. Decreased Bcl-2 expression and increased Bax/Bcl-2 ratio were also observed. The results suggest that reduced PGC1-α and IL-10, and elevated TLR4 proteins expression could be involved with the diminished mitochondrial biogenesis and anti-inflammatory response, as well as cell death signaling, in the establishment of hyperthyroidism-induced maladaptive cardiac hypertrophy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Hyperthyroidism is a disorder that causes many metabolic alterations, particularly in the cardiovascular system, which in the long term consists on deleterious cardiac remodeling, left ventricular fibrosis, and a decline in cardiac function (Dillmann 2010; Weltman et al. 2012). These alterations generate symptoms such as tachycardia, atrial fibrillation (5–10% of patients), increased systolic blood pressure, decreased diastolic pressure, and development of cardiac hypertrophy (Fazio et al. 2004).
The cardiac hypertrophy in hyperthyroidism is well described (Klein and Ojamaa 2001), and this heart remodeling can lead to increased diastolic dysfunction and cardiac dilatation, characteristics of heart failure (Li et al. 2007). One possible mechanism for the induction of hypertrophy in the early stages of hyperthyroidism may involve the modulation of peroxisome proliferator-activated receptor gamma coactivator (PGC1-α), that can interact with transcription factors involved in diverse types of biological responses (Liang and Ward 2006). PGC1-α can play a pivotal role in mitochondrial biogenesis and also serves as a marker of this process (Goldenthal et al. 2004; Ventura-Clapier et al. 2008). Important actions of this coactivator include the systemic regulation of lactate levels, a source of energy to the heart through the activation of lactate dehydrogenase (LDH), and the regulation of lipid metabolism through activation of pentose cycle (Summermatter et al. 2010, 2013; Messarah et al. 2011). The overexpression of PGC1-α tends to mitigate the cardiac hypertrophy induced by hyperthyroidism (Xu et al. 2012). In parallel, decreased PGC1-α expression can amplify the hypertrophic response, leading to maladaptive cardiac remodeling, and inducing cardiac dysfunction and inflammatory process (Garnier et al. 2003; Ventura-Clapier et al. 2008; Xu et al. 2012). Inflammatory cytokines decrease cardiac function through various mechanisms, influencing adrenergic signaling, calcium homeostasis, and redox signaling. These changes can lead to a vicious cycle involving the activation of pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukins (1β and 6), and ultimately provoking the stimulation of inflammatory signaling pathways activities (Khaper et al. 2010). In this scenario, toll-like receptors (TLRs) can be activated and stimulate the transduction of inflammatory information through intracellular proteins, such as MyD88 and nuclear factor kappa beta (NFκβ) (Darehgazani et al. 2016). On the other hand, to counter-regulate the inflammation, the intracellular HSP70 can inhibit inflammatory cytokines, such as TNF-α (Noble and Shen 2012). Nevertheless, extracellular HSP70 can also cause deleterious effects due to its capacity to interact with other molecules, such as toll-like receptors (2 and 4), inducing NF-κβ expression, and leading to an inflammatory response involving the expression of an intracellular adhesion molecule-1 (ICAM-1), interleukin 6 (IL-6), and keratinocyte-derived chemokine (KC) (Vondriska and Wang 2008). This inflammatory response promotes a decrease in cardiomyocyte contractility and can result in cell death, as well as heart failure (Satoh et al. 2006; Thuringer et al. 2015). Programmed cell death can be activated by the establishment of inflammatory processes. In this context, apoptosis signaling seems to be involved in the maladaptive hypertrophy through the modulation of proteins such as Bcl-2, Bax, p53 and caspases (caspase 3, 7 and 9), leading to DNA fragmentation and cell death (Harsdorf et al. 1999; Mihara et al. 1999; Narula et al. 1999; Chaudhari et al. 2009).
Therefore, the primary aim of this study was to investigate proteins involved in the inflammatory process, as well as the activation of cell death signaling in the maladaptive cardiac hypertrophy in the experimental hyperthyroidism.
2 Materials and methods
2.1 Ethical statement
This study was approved by the Ethics Committee on the use of animals at the Universidade Federal do Rio Grande do Sul (CEUA-UFRGS, protocol number 24504). It was conducted in accordance with the ethical principles established by the Universidade Federal do Rio Grande do Sul and the World Medical Association Declaration of Helsinki regarding the ethical conduct of research involving animals.
2.2 Experimental protocol
Male Wistar rats (169 ± 27 g, 34 days old, N=20) were obtained from the Center for Reproduction and Experimentation of Laboratory Animals, Universidade Federal do Rio Grande do Sul (CREAL-UFRGS). Animals were housed in plastic cages (four per cage) and received water and pelleted food ad libitum. They were maintained under standard laboratory conditions (controlled temperature of 21°C, 12 h light/dark cycle) and were divided into two groups: control (n=10); and hyperthyroid (T4, n=10).
T4 group rats received L- thyroxine (T4 – 12 mg/L) ad libitum in the drinking water for 28 days, as previously described (Araujo et al. 2011; da Rosa Araujo et al. 2010; Araujo et al. 2008). Animals in the control group received only vehicle (water). On the 28th day, the total body weight was measured and animals were anesthetized (ketamine 90mg/kg, xylazine 10mg/kg, I.P.) to collect blood in heparinized tubes (via the retro-orbital venous plexus, using a capillary tube filled with saline solution). The blood was later used for thyroid hormones analysis (free T3 and T4 in plasma). Animals were then euthanized (cervical dislocation), and heart and tibia were collected. The heart was weighed and then separated into left and right ventricle. The body weight, heart weight, and tibia length were used for the evaluation of the cardiac hypertrophy index. The left ventricle (LV) was homogenized and used to analyze the expression of proteins involved in the inflammatory and apoptotic signaling by western blot.
2.3 Thyroid hormones analysis
The blood samples were centrifuged for 15 minutes at 2415g in a refrigerated centrifuge at 4°C (Sorvall RC 5B - Rotor SM 24) to separate the plasma for the analysis of thyroid hormones. Free T3 and T4 were measured using chemiluminescence by competitive immunoassay, as previously described (Ferreira and Ávila 2013). The T3 levels were expressed in ng/mL and T4 levels in mg/dL.
2.4 Cardiac hypertrophy index evaluation
Body weight, heart weight, and tibia length were used to calculate two different cardiac hypertrophy indices. The cardiac hypertrophy index was calculated as the ratio between the total weight of heart tissue (in mg) and the tibia length (in cm) (Yin et al. 1982). A second calculation was performed by using the ratio of the total weight of heart tissue (in mg) to the total body weight (g) (Hu et al. 2003).
2.5 Western blot analysis
LV homogenization, electrophoresis, and protein transference were performed as previously described (Laemmli 1970). Total proteins in the homogenate were quantified (Lowry et al. 1951), using bovine albumin as a standard solution. The protein (50 µg) was submitted to one-dimensional sodium dodecyl sulfate-polyacrylamide gel electrophoresis in a discontinuous system using 8–12% (w/v) separating and stacking gels. The primary antibodies used for immunodetection were: HSP70 (70 kDa); HIF1-α (132 kDa); TLR4 (95 kDa); MyD88 (33 kDa); p-NFkβ (65kDa); NFkβ (65kDa); TNF-α (17kDa); IL-10 (20kDa); p-p38 (38kDa); p38 (38kDa); PGC1-α (90kDa); p53 (53kDa); Bax (20kDa); Bcl-2 (28kDa); Caspase-9 (17 kDa). Anti-rabbit, anti-mouse or anti-goat horseradish peroxidase-conjugate were used as secondary antibodies (Santa Cruz Biotechnology. Santa Cruz, CA). The protein bands were detected using chemiluminescence and the autoradiographs generated were quantitatively measured using the ImageJ software (public domain Java image processing program - National Institute of Mental Health, Bethesda, Maryland, USA). The molecular weights of the bands were determined by reference to a standard molecular weight marker (RPN 800 rainbow full range Bio-Rad, CA, USA). Results were normalized by the Ponceau method (Klein et al. 1995).
2.6 Statistical analysis
Results were expressed as the mean ± standard deviation (SD). Normality of data was tested by Kolmogorov-Smirnov homogeneity test. For comparisons between groups, Student’s t-test was used. Pearson’s test was used for correlation analysis. The significance level adopted was 5% (P<0.05). All calculations were performed using SPSS (Statistical Package for Social Sciences) version 20.0. Graph Pad Prism 5 was used to create the artwork.
3 Results
3.1 Thyroid hormones dosage
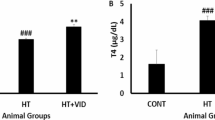
T4 levels were higher (310%) in the T4–treated group when compared to the control, indicating the development of hyperthyroidism in these animals. T3 levels, however, did not differ between groups. The T3/T4 ratio was lower (65%) in the T4-treated group, as compared to the control (table 1).
3.2 Cardiac hypertrophy analysis
The total heart weight was significantly increased (14%) in the T4 group as compared to control. When compared to control, the T4 group showed higher values for the two hypertrophy indices: heart in relation to body weight (20%), and heart/tibia length (14%). These results are summarized in table 1.
3.3 Western blot analysis of LV homogenate
Western blot analysis was conducted to verify the expression of proteins involved in inflammatory and apoptotic signaling. The expression of HSP70 (figure 1A) and HIF1-α (figure 1B) were not significantly different between the groups. TLR4 was increased (112%) in the T4 group compared to the control group (figure 2A); however, no changes in MyD88 (figure 2B), p-NFκB (figure 2C), NFκB (figure 2D) and TNF- α (figure 3A) expression were detected between the two groups. Nevertheless, there was a decrease in IL-10 expression in the T4 group (figure 3B); but the IL-10/TNF-α ratio was not changed (figure 3C). The expression of p-p38 (figure 4A) and p38 (figure 4B) was not altered; however, PGC1-α (figure 4C) and p53 (figure 4D) showed significantly lower levels in the T4 group (73% and 61%, respectively) as compared to the control group. There was a negative correlation between PGC1-α and TLR4 (r=−.686, n=9, p=0.041).

Representative image bands and respective graphs of (A) HSP70 and (B) HIF1-α in control and T4 groups. Values represent mean ± SD. n = 4 animals in control group and 5 animals in the T4 group. Bands were normalized using the Ponceau method and data were analyzed by Student’s t-test.

Representative image bands and respective graphs of (A) TLR4, (B) MyD88, (C) p-NFκB, and (D) NFκB in control and T4 groups. Values represent mean ± SD. n = 4 animals in control group and 5 animals in the T4 group. Bands were normalized using the Ponceau method and data were analyzed by Student’s t-test.

Representative image bands and respective graphs of (A) TNF-α, (B) IL-10, and (C) IL-10/TNF-α ratio in control and T4 groups. Values represent mean ± SD. n = 4 animals in control group and 5 animals in the T4 group. Bands were normalized using the Ponceau method and data were analyzed by Student’s t-test. * P<0.05 compared to control group.

Representative image bands and respective graphs of (A) p-p38, (B) p38, (C) PGC1-α, and (D) p53 in control and T4 groups. Values represent mean ± SD. n = 4 animals in control group and 5 animals in the T4 group. Bands were normalized using the Ponceau method and data were analyzed by Student’s t-test. * P<0.05 compared to control group. ** P<0.001 compared to control group.
Bax levels were not different between the groups (figure 5A); however, Bcl-2 (figure 5B) showed diminished levels (65%) in the T4 group as compared to the control group. The Bax/Bcl-2 ratio was higher (60%) in the T4 group compared to control (figure 5C). Caspase-9 expression was not different between the groups (figure 5D).

Representative image bands and respective graphs of (A) Bax, (B) Bcl-2, (C) Bax/Bcl-2 ratio, and (D) caspase-9 in control and T4 groups. Values represent mean ± SD. n = 4 animals in control group and 5 animals in the T4 group. Bands were normalized using the Ponceau method and data were analyzed by Student’s t-test. * P<0.05 compared to control group. ** P<0.001 compared to control group.
4 Discussion
In this study, the main finding was an association between the cardiac hypertrophy and the alterations of metabolic regulators, given by reduced PGC1-α and p53 expression in the hyperthyroidism. In addition, an activation of the inflammatory signaling, with elevated TLR4 and diminished IL-10 expression was also observed in this model. Furthermore, this inflammation process seems to be associated with a reduction in Bcl-2 expression and augmented Bax/Bcl-2 ratio, indicating an intracellular pro-apoptotic signaling activation.
Development of hyperthyroidism was verified as in previous studies, by evaluating free T4 levels in plasma and cardiac hypertrophy indices (Messarah et al. 2011; Araujo et al. 2007, 2008, 2011). Increased T4 levels were found in the plasma of hyperthyroid group. Although no statistically significant, T3 levels also increased by 54% in the hyperthyroid group. T3 and T4 increase were accompanied by the development of cardiac hypertrophy, a classical evidence that indicates the development of thyrotoxicosis in these animals (Xu et al. 2012). As previously documented, thyroid hormones are responsible for various hyperthyroidism–induced cardiovascular changes that can lead to heart mass increment (Fazio et al. 2004; Li et al. 2007; Dillmann 2010; Weltman et al. 2012; Thuringer et al. 2015). The cardiac hypertrophy in the present study was confirmed through all cardiac hypertrophy indices, which were higher in the T4 group than in control. As well as observed in this study, previous data have also found hyperthyroidism-induced cardiac hypertrophy (Araujo et al. 2006, 2007, 2008, 2011). However, the adaptive cardiac hypertrophy could advance to maladaptive hypertrophy and progress to heart failure (Dillmann 2010; Xu et al. 2012). The transition between these cardiac hypertrophic stages may involve a detrimental condition of energetic metabolism. In this scenario, factors involved in mitochondrial transcription such as PGC1-α play a pivotal role in metabolic control.
The augmented PGC1-α expression in cell stimulates the mitochondrial biogenesis, activation of genes involved in the control of oxidative phosphorylation, such as nuclear respiratory factor (NFR), estrogen-related receptor (ERR), peroxisome proliferator-actived receptor (PPARs), and myocyte enhancer factor -2 (MEF2) (Tuomainen and Tavi 2017). On the other hand, PGC1-α seems to be related with the control of cardiac hypertrophy in both adaptive and maladaptive conditions (Goldenthal et al. 2004; Narula et al. 1999; Ventura-Clapier et al. 2008; Messarah et al. 2011; Summermatter et al. 2010, 2013; Xu et al. 2012). In our study, PGC1-α protein expression was reduced in rats with hyperthyroidism (figure 4C). This result agrees with previous studies, in which it was described that hyperthyroidism induced the reduction of PGC1-α expression. However, in such studies, the thyroid hormones were administered in short term as compared to our one (Xu et al. 2012; Maity et al. 2013). Such decreased PGC1-α levels, in long term, could be associated with maladaptive hypertrophy, since this reduction in cardiac muscle provokes relevant mitochondrial dysfunction and consequent cardiac phenotype changes and potential progression to heart failure (Ventura-Clapier et al. 2008; Maity et al. 2013). All these actions are mediated by PGC1-α, demonstrating its key role as a cell regulator of energetic metabolism in this model. This relevant aspect stands out, since other metabolic signaling factors remained unaltered in T4–treated rats, such as HSP70 and HIF1-α (figure 1A-B), as previously reported in hyperthyroid animals (Rodriguéz-Goméz et al. 2013). Additionally, as it was previously demonstrated, decreases in PGC1-α expression in the heart failure are strongly associated with inflammatory and apoptotic alterations, which were also observed in our study (Schilling et al. 2011; Sen et al. 2011; Maity et al. 2013; Darehgazani et al. 2016).
In this context, IL-10 levels were lower in T4 group as compared to the control group (figure 3B), indicating a possibly reduced anti-inflammatory potential in hyperthyroid animals, since IL-10 is involved in tissue repair and therefore considered to provide beneficial effects on cardiac function in disease states (Bartekova et al. 2018). In addition, the hyperthyroid animals also demonstrated increased TLR4 levels (figure 2A), a signaling protein involved in inflammation. TLR4 may induce inflammation in pathological conditions and was previously associated with depression of myocardial function, maladaptive left ventricular remodeling and progression of cardiovascular diseases (Feng and Chao 2011; Vallejo 2011; Timmers et al. 2008; Jia et al. 2014; Vaez et al. 2016; Yang et al. 2016). In the inflammatory process, the activation of TLR4 can be suppressed by the action of peroxisome proliferator-activated receptor-γ (PPARγ) (Darehgazani et al. 2016). Since PPARγ is co-activated by PGC1-α, it can be suggested that diminished PGC1-α expression provide an inadequate PPARγ activation, increasing TLR4 expression, which would stimulate the inflammatory process and suppress the anti-inflammatory response through reduced IL-10 expression (Puigserver 2005). In fact, this hypothesis is supported by a previous study, which demonstrated that TLR4 activation is directly related with downregulation of PGC1-α in the heart failure (Schilling et al. 2011).
The maladaptive cardiac hypertrophy may involve an imbalance of factors that regulate the cell cycle, for example, p53. This protein is known as a cell development modulator, controlling the expression of genes responsible for the metabolism (Sen et al. 2011). Interestingly, the results showed a reduction in the protein expression of p53 in the T4 group (figure 4D), which was associated with diminished PGC1-α levels. It has been reported that PGC1-α can also modulate the activity of p53 (Sen et al. 2011). On the other hand, this result could explain the conservation of NFkB concentration, since increased p53 levels could block NFkB gene expression (Dijsselbloem et al. 2007; Madenspacher et al. 2013; Olivetti et al. 1997). Although a reduction in p53 expression was observed, this result was not enough in order to elevate NFkB expression. However, the decrease in the expression of IL-10, in conjunction with the elevation in TLR4 observed in our study, could be associated with a possible increase in the inflammatory response, and consequently cell death activation, an important path to progression from adaptive to maladaptive hypertrophy.
Regarding cell survival/death, caspase-9, a protein involved in apoptosis signaling, remained unaltered in T4–treated mice. However, although Bax expression was not different between groups, Bcl-2 expression showed a reduction in the T4 group (figure 5A). Since Bcl-2 is a cell survival signaling protein, its reduction could indicate a suppression of cell survival in T4–treated animals. As previously described, when Bcl-2 is available it can form heterodimers with Bax and promote cell survival; however, a predominance of Bax homodimers (as would be evident with low Bcl-2 expression) promotes cell death (Olivetti et al. 1997). The Bax/Bcl-2 ratio was increased in the T4 group, suggesting a propensity towards apoptosis in these animals.
In summary, a reduction in PGC1-α and p53 expression in hyperthyroidism reflects a more advanced disease stage, which could lead to the establishment of maladaptive cardiac hypertrophy and possible development of heart failure. In this context, the TLR4 protein expression was correlated with downregulation of PGC1-α, indicating that the activation of inflammatory proteins is associated with reduced mitochondrial biogenesis and posterior apoptotic factors activation, which can have a detrimental role in this hypertrophy stage.
References
Araujo ASR, Diniz GP, Seibel FER, Branchini G, Ribeiro MFM, Brum IS, Khaper N, Barreto-Chaves ML and Belló-Klein A 2011 Reactive oxygen and nitrogen species balance in the determination of thyroid hormones-induced cardiac hypertrophy mediated by renin–angiotensin system. Mol. Cell. Endocrinol. 333 78–84
Araujo ASR, Enzveiler AT, Schenkel P, Fernandes TRG, Ribeiro MFM, Partata WA, Llesuy S and Belló-Klein A 2007 Oxidative stress activates insulin-like growth factor I receptor protein expression, mediating cardiac hypertrophy induced by thyroxine. Mol. Cell. Biochem. 303 89–95
Araujo ASR, Ribeiro MFM, Enzveiler A, Schenkel P, Fernandes TRG, Partata WA, Irigoyen MC, Llesuy S and Belló-Klein A 2006 Myocardial antioxidant enzyme activities and concentration and glutathione metabolism in experimental hyperthyroidism. Mol. Cell. Endocrinol. 249 133–139
Araujo ASR, Schenkel P, Enzveiler AT, Fernandes TRG, Partata WA, Llesuy S, Ribeiro MF, Khaper N, Singal PK and Belló-Klein A 2008 The role of redox signaling in cardiac hypertrophy induced by experimental hyperthyroidism. J. Mol. Endocrinol. 41 423–430
Bartekova M, Radosinska J, Jelemensky M and Dhalla NS 2018 Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. [Epub ahead of print] doi: 10.1007/s10741-018-9716-x.
Chaudhari M, Jayaraj R, Bhaskar ASB and Lakshmana Rao PV 2009 Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 262 153–161
da Rosa Araujo AS, Fernandes T, Ribeiro MF, Khaper N and Belló-Klein A 2010 Redox Regulation of Myocardial Erk 1/2 Phosphorylation in Experimental Hyperthyroidism: Role of Thioredoxin-Peroxiredoxin System. J. Cardiovasc. Pharmacol. 56 513–517
Darehgazani R, Peymani M, Hashemi M-S, Omrani MD, Movafagh A, Ghaedi K and Nasr-Esfahani MH 2016 PPARγ ameliorated LPS induced inflammation of HEK cell line expressing both human Toll-like receptor 4 (TLR4) and MD2. Cytotechnology 68 1337–148
Dijsselbloem N, Goriely S, Albarani V, Gerlo S, Francoz S, Marine J-C, Goldman M, Haegeman G and Vanden Berghe W 2007 A critical role for p53 in the control of NF-kappaB-dependent gene expression in TLR4-stimulated dendritic cells exposed to Genistein. J. Immunol. 178 5048–5057
Dillmann W 2010 Cardiac hypertrophy and thyroid hormone signaling. Heart Fail. Rev. 15 125–132
Fazio S, Palmieri EA, Lombardi G and Biondi B 2004 Effects of thyroid hormone on the cardiovascular system. Recent Prog. Horm. Res. 59 31–50.
Feng Y and Chao W 2011 Toll-like receptors and myocardial inflammation. Int. J. Inflam. 2011 170352
Ferreira A and Ávila S 2013 Diagnóstico Laboratorial das Principais Doenças Infecciosas e Auto-imunes. 2nd ed. (Rio de Janeiro: Guanabara Koogan).
Garnier A, Fortin D, Deloménie C, Momken I, Veksler V and Ventura-Clapier R 2003 Depressed mitochondrial transcription factors and oxidative capacity in rat failing cardiac and skeletal muscles. J. Physiol. 551 491–501
Goldenthal MJ, Weiss HR and Marín-García J 2004 Bioenergetic remodeling of heart mitochondria by thyroid hormone. Mol. Cell. Biochem. 265 97–106
Harsdorf VR, Li PF and Dietz R 1999 Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99 2934–2941
Hu LW, Benvenuti LA, Liberti EA, Carneiro-Ramos MS and Barreto-Chaves MLM 2003 Thyroxine-induced cardiac hypertrophy: influence of adrenergic nervous system versus renin-angiotensin system on myocyte remodeling. Am. J. Physiol. Integr. Comp. Physiol. 285 R1473–R1480
Jia S-J, Niu P-P, Cong J-Z, Zhang B-K and Zhao M 2014 TLR4 signaling: A potential therapeutic target in ischemic coronary artery disease. Int. Immunopharmacol. 23 54–59
Khaper N, Bryan S, Dhingra S, Singal R, Bajaj A, Pathak CM and Singal PK 2010 Targeting the vicious inflammation–oxidative stress cycle for the management of heart failure. Antioxid. Redox. Signal. 13 1033–1049
Klein D, Kern RM and Sokol RZ 1995 A method for quantification and correction of proteins after transfer to immobilization membranes. Biochem. Mol. Biol. Int. 36 59–66
Klein I and Ojamaa K 2001 Thyroid hormone and the cardiovascular system. New Engl. J. Med. 344 501–509
Laemmli UK 1970 Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227 680–685
Li H, Yin H, Yao Y, Shen B, Bader M, Chao L and Chao J 2007 Tissue kallikrein protects against pressure overload-induced cardiac hypertrophy through kinin B2 receptor and glycogen synthase kinase-3β activation. Cardiovasc. Res. 73 130–142
Liang H and Ward WF 2006 PGC-1α: a key regulator of energy metabolism. Adv. Physiol. Educ. 30 145–151
Lowry OH, Rosebrough NJ, Farr AL and Randall RJ 1951 Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193 265–275
Madenspacher JH, Azzam KM, Gowdy KM, Malcolm KC, Nick JA, Dixon D, Aloor JJ, Draper DW, Guardiola JJ, Shatz M, Menendez D, Lowe J, Lu J, Bushel P, Li L, Merrick BA, Resnick MA and Fessler MB 2013 p53 integrates host defense and cell fate during bacterial pneumonia. J. Exp. Med. 210 891–904
Maity S, Kar D, De K, Chander V and Bandyopadhyay A 2013 Hyperthyroidism causes cardiac dysfunction by mitochondrial impairment and energy depletion. J. Endocrinol. 217 215–228
Messarah M, Saoudi M, Boumendjel A, Boulakoud MS and Feki A El 2011 Oxidative stress induced by thyroid dysfunction in rat erythrocytes and heart. Environ. Toxicol. Pharmacol. 31 33–41
Mihara S, Suzuki N, Wakisaka S, Suzuki S, Sekita N, Yamamoto S, Saito N, Hoshino T and Sakane T 1999 Effects of thyroid hormones on apoptotic cell death of human lymphocytes. J. Clin. Endocrinol. Metab. 84 1378–1385
Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, Bello Bd, Semigran MJ, Bielsa-Masdeu A, Dec Gw, Israels S, Ballester M, Virmanii R, Saxena S and Kharbanda S 1999 Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc. Natl. Acad. Sci. U S A 96 8144–8149
Noble EG and Shen GX 2012 Impact of exercise and metabolic disorders on heat shock proteins and vascular inflammation. Autoimmune. Dis. 2012 836519
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, Quaini E, Loreto CD, Beltrami CA, Krajewski S, Reed JC and Anversa P 1997 Apoptosis in the failing human heart. New Engl. J. Med. 336 1131–1141
Puigserver P 2005 Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-α. Int. J. Obes. 29 S5–S9
Rodriguéz-Goméz I, Banegas I, Wangensteen R, Quesada A, Jiménez R, Gómez-Morales M, O’Valle F, Duarte J and Vargas F 2013 Influence of thyroid state on cardiac and renal capillary density and glomerular morphology in rats. J. Endocrinol. 216 43–51
Satoh M, Shimoda Y, Akatsu T, Ishikawa Y, Minami Y and Nakamura M 2006 Elevated circulating levels of heat shock protein 70 are related to systemic inflammatory reaction through monocyte Toll signal in patients with heart failure after acute myocardial infarction. Eur. J. Heart Fail. 8 810–815
Schilling J, Lai L, Sambandam N, Dey CE, Leone TC and Kelly DP 2011 Toll-like receptor-mediated inflammatory signaling reprograms cardiac energy metabolism by repressing peroxisome proliferator-activated receptor coactivator-1 signaling. Circ. Heart Fail. 4 474–482
Sen N, Satija YK and Das S 2011 PGC-1α, a Key Modulator of p53, promotes cell survival upon metabolic stress. Mol. Cell. 44 621–634
Summermatter S, Baum O, Santos G, Hoppeler H and Handschin C 2010 Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J Biol. Chem. 285 32793–32800
Summermatter S, Santos G, Perez-Schindler J and Handschin C 2013 Skeletal muscle PGC-1 controls whole-body lactate homeostasis through estrogen-related receptor-dependent activation of LDH B and repression of LDH A. Proc. Natl. Acad. Sci. 110 8738–8743
Thuringer D, Berthenet K, Cronier L, Jego G, Solary E and Garrido C 2015 Oncogenic extracellular HSP70 disrupts the gap-junctional coupling between capillary cells. Oncotarget 6 10267–10283
Timmers L, Sluijter JPG, van Keulen JK, Hoefer IE, Nederhoff MGJ, Goumans M-J, Doevendans PA, van Echteld CJ, Joles JA, Quax PH, Piek JJ, Pasterkamp G and de Kleijn DP 2008 Toll-Like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ. Res. 102 257–264
Tuomainen T and Tavi P 2017 The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp. Cell. Res. 360 12–18
Vaez H, Najafi M, Rameshrad M, Toutounchi NS, Garjani M, Barar J, Garjani A 2016 AMPK activation by metformin inhibits local innate immune responses in the isolated rat heart by suppression of TLR 4-related pathway. Int. Immunopharmacol. 40 501–507
Vallejo JG 2011 Role of Toll-like receptors in cardiovascular diseases. Clin. Sci. 121 1–10
Ventura-Clapier R, Garnier A and Veksler V 2008 Transcriptional control of mitochondrial biogenesis: the central role of PGC-1. Cardiovasc. Res. 79 208–217
Vondriska TM and Wang Y 2008 A new (heat) shocking player in cardiac hypertrophy. Circ. Res. 103 1194–1196
Weltman NY, Wang D, Redetzke RA and Gerdes AM 2012 Longstanding hyperthyroidism is associated with normal or enhanced intrinsic cardiomyocyte function despite decline in global cardiac function. PLoS One 7 e46655
Xu W, Hou D, Jiang X, Lu Z, Guo T, Liu Y, Wang D, Zen K, Yu B and Zhang CY 2012 The protective role of peroxisome proliferator-activated receptor γ coactivator-1α in hyperthyroid cardiac hypertrophy. J Cell. Physiol. 227 3243–3253
Yang Y, Lv J, Jiang S, Ma Z, Wang D, Hu W, Deng C, Fan C, Di S, Sun Y and Yi W 2016 The emerging role of Toll-like receptor 4 in myocardial inflammation. Cell Death. Dis. 7 e2234–e2234
Yin FC, Spurgeon HA, Rakusan K, Weisfeldt ML and Lakatta EG 1982 Use of tibial length to quantify cardiac hypertrophy: application in the aging rat. Am. J Physiol. 243 H941–H947
Acknowledgments
Veterinary medical support from Dr. André Ricardo Ribeiro Belló is acknowledged. We also acknowledge Professor Ilma Simoni Brum, Physiology Department, UFRGS, for providing the p53 antibody. This paper was written during a scholarship provided by the International Cooperation Program CAPES/DFATD at the University of Manitoba. This study was funded by Conselho Nacional de Desenvolvimento Científico e tecnológico, Fundação de Amparo à Pesquisa do Estado do Rio Grande do Sul and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Seyed E Hasnain.
Corresponding editor: Seyed E Hasnain
Rights and permissions
About this article

Cite this article
Teixeira, R.B., Barboza, T.E., de Araújo, C.C. et al. Decreased PGC1-α levels and increased apoptotic protein signaling are associated with the maladaptive cardiac hypertrophy in hyperthyroidism. J Biosci 43, 887–895 (2018). https://doi.org/10.1007/s12038-018-9816-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12038-018-9816-8