
Abstract
The trans-sulfuration pathway is a biochemical mechanism that links methionine metabolism to the biosynthesis of cellular redox-controlling molecules, like cysteine, glutathione, and taurine. While there is some knowledge about the metabolic intermediates and enzymes that participate in trans-sulfuration, little is known about the physiological importance of this mechanism. Deficiencies within the trans-sulfuration pathway induces (i) the generation of reactive species of oxygen (ROS) and halogens (RHS), (ii) homocyst(e)ine accumulation, and (iii) the synthesis of proinflammatory molecules by macrophages, and contribute to humans pathologies like atherosclerosis and tumor development. In this review we outline the role of this biochemical pathway in tumor development and analyze current findings on the role of trans-sulfuration in mammalian physiology. The potential relationship between chronic inflammation, and tumor and atherosclerotic development are discussed.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Methionine is an essential amino acid that influences cellular metabolism. It is a proteinogenic amino acid and an essential precursor for the synthesis of glutathione (γ-glutamyl-cysteinyl-glycine; GSH), a molecule that participates in spermine and spermidine synthesis and is the major source of methyl groups for biological molecules. Methionine is also necessary for cysteine biosynthesis, a process carried out through a specialized biochemical pathway called the trans-sulfuration pathway [1]. Methionine metabolites are essential for phosphatidyl choline regeneration in the cytoplasmic membrane, and methionine is required for the metabolic pathways that regulate gene expression, chromatin structure, transcription, post-transcriptional processing, and protein synthesis [2].
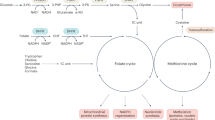
Methionine metabolism can be divided into five major but interdependent biochemical pathways: (i) the methionine salvage pathway, (ii) the methionine cycle, (iii) the trans-sulfuration pathway, (iv) the GSH synthesis pathway, and (v) the taurine synthesis pathway (Fig. 1) [1, 3, 4]. The methionine salvage pathway is important for methionine conservation in cells that synthesize large amounts of polyamines for the cell cycle [1]. In the salvage pathway, methylthioadenosine phosphorylase (MTAP; E.C. 2.4.2.28) (Fig. 1, reaction 14) is one of the key enzymes. Deficiencies in the salvage pathway are responsible for many kinds of tumors, including non-small cell lung cancer, leukemia, glioma, rectal adenocarcinoma, and melanoma [5–8].

A simplified metabolic chart of biologically important sulfur compounds. The five discrete, interconnected pathways are indicated by different shades of gray. The enzymes required in each pathway are (1) methionine adenosyltransferase (MAT), (2) general methyltransferase, (3) adenosylhomocysteinase, (4) cystathionine β-synthase, (5) γ-cystathionase, (6) γ-glutamilcysteine synthase, (7) glutathione synthase, (8) choline dehydrogenase plus betaine aldehyde dehydrogenase, (9) betaine homocysteine methyltransferase, (10) methionine synthase, (11) serine-hydroxymethylase, (12) methylenetetrahydrofolate reductase, (13) spermidine synthase, (14) methylthioadenosine phosphorylase, (15) S-methyl-5-thioribose-1-phosphate isomerase, (16) methylthioribulose 1-phosphate dehydratase, (17) tyrosine transaminase, and (18) cysteine dioxygenase
The methionine cycle plays an important role in cell physiology in a broad sense. It is the place where S-adenosylmethionine (SAM) biosynthesis occurs in a reaction catalyzed by methionine adenosyltransferase (MAT, EC 2.5.1.6; Fig. 1, reaction 1) [9]. SAM is the major biological methyl donor for other molecules such as DNA and plays an important allosteric role in the regulation of sulfur metabolism [9]. The methionine cycle is also responsible for (i) converting betaine into dimethyl-glicine, which regenerates methionine from cysteine in a reaction catalyzed by betaine homocysteine methyltransferase (EC 2.1.1.5; Fig. 1, reaction 8), and (ii) converting 5-methylenetetrahydrofolate (5-THF) into tetrahydrofolate (THF; Fig. 1, reaction 10) by methionine synthase (EC 2.1.1.13), which produces methionine from homocysteine (Fig. 1) [3, 10].
The biochemical mechanism of trans-sulfuration is less well characterized than the methionine salvage and cycle pathways. However, the trans-sulfuration pathway is necessary for cellular physiology because it connects the methionine metabolic pathways to the generation of cysteine, glutathione (GSH), and taurine (Fig. 1) [11, 12]. Tarver et al. first demonstrated that cysteine could be synthesized from methionine in 1939 [12], and called it the trans-sulfuration or cystathionine pathway [11]. This pioneer work served as the base for further characterization of the trans-sulfuration pathway using isolated hepatocytes from the mammalian liver [13]. The first step of trans-sulfuration involves the formation of cystathionine from homocysteine and serine, in a reaction catalyzed by cystathionine synthase (EC 2.5.1.48; Fig. 1, reaction 4). Once formed, cystathionine is cleaved by cystathionase (cystathionine γ-lyase; EC 4.4.1.1), releasing free cysteine (Fig. 1, reaction 5). In the presence of high cysteine levels, cystathionine is directed into the GSH and taurine synthesis pathways [14]. Thus, cysteine levels are considered the limiting step in liver GSH synthesis [11, 14, 15], where approximately 50% of the cysteine used for GSH anabolism is derived from methionine that was synthesized from the trans-sulfuration pathway [16, 17]. When homocysteine levels are low, cysteine flux through the trans-sulfuration pathway becomes down-regulated in order to conserve homocysteine for the methionine cycle [18]. Trans-sulfuration activity in the liver is markedly impaired or absent in fetuses, newborns, cirrhotic patients, and patients with homocyst(e)inemia [19]. The trans-sulfuration pathway is also blocked in some liver tumor cell lines, including HepG2 and HuH-7 cells [20]. While the mechanism of this blockade is currently unknown, these cells are unable to form GSH from methionine but can synthesize large amounts of homocyst(e)ine [20]. It is demonstrated that approximately 50% of all tumors are incapable of proliferating when methionine is replaced by homocysteine, resulting in cell cycle arrest, strict inhibition of mitosis, and eventual death [21–26]. Tumor cells export large amounts of homocyst(e)ine when placed in culture medium that contains high methionine, suggesting that there is an anomaly in methionine metabolism [2]. This absolute requirement for methionine by tumor cells is known as “methionine dependency” or “methionine stress” [1, 2]. The molecular mechanism of methionine dependency is currently unknown, but may be triggered by high transmethylation rates [27] that result in the hyperproduction of S-adenosylhomocysteine (AdoHcy) and homocysteine [1]. While the influence of methionine-dependence on trans-sulfuration is unknown, the accumulation and export of homocysteine probably impairs cysteine biosynthesis possibly affecting the function of major enzymes in the trans-sulfuration pathway. An understanding of how the major enzymes of the trans-sulfuration pathway control the flux of sulfur-containing molecules and cellular redox homeostasis is necessary in order to understand the role of this biochemical mechanism in tumor growth and development.
The first enzyme of the trans-sulfuration pathway: adenosylhomocysteinase
Adenosylhomocysteinase (S-Adenosylhomocysteine hydrolase or AdoHcyase; EC 3.3.1.1) is a cytoplasmic enzyme that catalyzes the reversible hydrolysis of AdoHcy into adenosine and homocysteine (Fig. 1, reaction 3) [28, 29]. AdoHcyase is a tetrameric protein with a molecular weight of 45–50 kDa, in which each subunit contains one molecule of tightly bound NAD+ [28, 30], as determined by X-ray crystallography (Fig. 2) [31, 32]. Three-dimensional characterization of mammalian AdoHcyase showed that the four NAD+-binding domains are located near the center of the tetramer while the catalytic domains are located far from the center [32].

Three-dimensional models of tetrameric human adenosyl-homocysteinase (Protein Data Bank accession number 1LI4), cystathionine β-synthase (Protein Data Bank accession number 1JBQ), and monomeric yeast γ-cystathionase (Protein Data Bank accession number 1N8P). A pyridoxal-5′-phosphate molecule is shown in the active site of yeast γ-cystathionase. The models are shown at a 90° angle to illustrate their distinctive characteristics
The thermodynamic equilibrium of the reaction favors AdoHcy synthesis in vitro [33], but the hydrolysis of AdoHcy into adenosine and homocysteine by AdoHcyase prevails under physiological conditions because both reaction products are rapidly removed [29]. Once formed, adenosine is either deaminated by adenosine deaminase (EC 3.5.4.4) or enters the purine nucleotide pool through the action of adenosine kinase (EC 2.7.1.20). Homocysteine can enter the trans-sulfuration pathway and be metabolized into cystathionine (Fig. 1, reaction 4) or be re-methylated into methionine (Fig. 1, reactions 8 and 10) [29]. The existence of multiple routes for the metabolism of AdoHcy (SAM) precursors and its metabolic products, adenosine and homocysteine, is consistent with the requirement for efficient product removal to prevent AdoHcy accumulation. AdoHcy accumulation can interfere with AdoMet-dependent methyltransferases by product inhibition, which results in epigenetic alterations [29]. The AHCY gene, which encodes AdoHcyase, was mapped to the human chromosome 20q13.1 [34], and many alleles are associated with hypermethioninemia and coronary heart disease [35, 36].
The second enzyme of the trans-sulfuration pathway: cystathionine β-synthase
The cystathionine β-synthase (EC 4.2.1.22; CBS) gene, situated on chromosome 21q22.3, encodes an enzyme that catalyzes the synthesis of cystathione, the second step in the trans-sulfuration pathway (Fig. 1, reaction 4) [37]. Human cystathionine β-synthase is a 63 kDa enzyme that contains an N-terminal heme domain that regulates enzymatic activity, a catalytic domain that binds PLP (pyridoxal 5′-phosphate), and a C-terminal domain that confers responsiveness to the allosteric activator SAM. The full-length human enzyme exists in a tetrameric state (Fig. 2) [38], in which the catalytic core resembles other members of the fold II family of PLP-dependent enzymes [39]. Thus, because of its location at a branch point for remethylation and trans-sulfuration, cystathionine β-synthase is a highly regulated enzyme (Fig. 1, reaction 4) [4]. The C-terminal regulatory domain inhibits activity of the full-length enzyme, but binding of SAM to this domain converts the enzyme to an activated state [40]. The activation of full-length cystathionine β-synthase by SAM involves a conformational change, so that the C-terminal domain can no longer play an autoinhibitory role [38]. Interestingly, point mutations in the C-terminal domain of cystathionine β-synthase have been identified in homocyst(e)inuric patients [4], and shown to result in diminished SAM responsiveness and varying degrees of constitutive activation [41, 42]. The consequence of cystathionine β-synthase activation by SAM is an increase in the remotion of homocysteine through the trans-sulfuration pathway. The heme group present in the N-terminus of mammalian cystathionine β-synthase may act as a redox sensor [39]. While reduction of the heme moiety is associated with decreased enzyme activity, flux through cystathionine β-synthase is increased under oxidizing conditions [43]. Redox regulation of cystathionine β-synthase activity is consistent with the reported reciprocal sensitivity of the trans-sulfuration pathway to pro- and antioxidants, which enhance or diminish homocysteine flux through the trans-sulfuration pathway, respectively [43].
Cystathionine β-synthase enzyme deficiencies are associated with mental retardation, elevated homocysteine levels [homocyst(e)inuria], skeletal abnormalities, elevated risk of blood clots and atherosclerosis [44], as well as neurodegenerative diseases like Alzheimers and dementia [45]. Homocyst(e)inuria is correlated with other vascular effects like atherothrombosis and endothelial dysfunction because of its auto-oxidative potential, and a related increase in the production of reactive oxygen species (ROS) [46]. Since reduced free homocysteine contains a free thiol group, this molecule can react with itself to form homocystine, or with albumin and cysteine to increase RS production [47]. Plasma levels of reduced free homocysteine are found to enhance oxidative stress [47]. In addition, chronic exposure of endothelial cells to homocysteine accelerates the rate of endothelial senescence and decreases telomere length for every population doubling [48].
The third enzyme of the trans-sulfuration pathway: γ-cystathionase
γ-Cystathionase (CSE, cystathionine γ-lyase; EC 4.4.1.1) is a PLP-dependent enzyme that catalyzes the conversion of cystathionine into cysteine, α-ketobutyrate, and ammonia (Fig. 1, reaction 5). l-cysteine is further metabolized in the liver to yield GSH or taurine, although CSE itself is capable of metabolizing cyst(e)ine and producing H2S, a gaseous neuromodulator and smooth-muscle relaxant [49].
Messerschmidt et al. resolved the three-dimensional structure of yeast CSE and showed that it exists in a tetrameric state [50]. Each monomer contains one PLP cofactor and 393 amino acids (Fig. 2). Similar to other PLP-dependent enzymes, the monomer comprises three structurally and functionally distinct regions in which the N-terminus interacts with the active site of the neighboring monomer and a large PLP-binding domain that carries most of the catalytically important residues [50].
The CTH gene on human chromosome 1p31.1 encodes the enzyme CSE [51]. Recessive mutant alleles for CTH are associated with cystathioninuria, a disease characterized by abnormal accumulation of plasma that leads to increased urinary excretion of cystathionine [51]. Screening surveys of neonatal urine samples estimated the prevalence of cystathioninuria at ∼1:14,000 live births [52, 53].
The trans-sulfuration pathway and tumor protection/promotion: is there a link?
The three enzymatic steps of the trans-sulfuration pathway provide the amount of cysteine required to synthesize the cellular redox-controlling molecules, like GSH and taurine, that protect the molecular constituents of cells against reactive species (SP)-induced damages (Fig. 3A). For example, mitochondrial and nuclear DNA are continuously and chemically damaged by both endogenous and exogenous RS, which contributes to degenerative processes such as aging and cancer [54]. It is widely accepted that RS produce a broad range of DNA damage including base and sugar modifications, base-free sites, DNA-protein cross-links, and strand breaks [55–58]. Until now, more than 20 different types of base damage have been identified following exposure to oxidative stress [59]. The most prevalent damage to purines is 7,8-dihydro-8-oxoguanine (8-oxoguanine or 8-oxoG), while the most common damage to pyrimidines is the formation of thymine glycol (Tg) [59]. Modified bases generated by RS are highly mutagenic and can induce base mispairing during DNA replication, generating mutations that affect cellular physiology [59].

A model showing how tumors can form from deficiencies in the trans-sulfuration pathway. The equilibrium between methionine homeostasis and the trans-sulfuration pathway balances the level of homocysteine in cells and keeps the redox homeostasis (A). Metabolic deficiencies in the trans-sulfuration pathway imbalance homocysteine levels in the cell, which increases homocysteine concentration and alters redox homeostasis, as well as the DNA methylation pattern. The low concentration of GSH and taurine leads to an increase in reactive oxygen species (ROS) generation, DNA damage, gene mutation, protein inactivation, and the altered activity of cell cycle checkpoints. All these cellular phenomena may ultimately result in tumor growth and expansion (B)
Oxidative stress and carcinogenesis
The specific mechanism by which oxidative stress contributes to carcinogenesis is largely unknown [60], but two different mechanisms are thought to play a role: (i) modulation of gene expression and (ii) induction of genetic modifications [60]. Modulation of gene expression by oxidative damage affects carcinogenesis by altering the epigenetic effects and chromosomal rearrangements. Epigenetic effects on gene expression stimulate growth signals and proliferation [60], while chromosomal rearrangements contribute to neoplasic progression [60]. In addition, a redox imbalance is shown to stimulate protein kinase and poly(ADP ribosylation) pathways, affecting signal transduction and promoting tumor development [61]. Interestingly, studies examining the effects of high homocysteine levels show that poly(ADP-ribose) polymerase (PARP) enzymes repair single strand DNA breaks (SSBs) induced by genotoxic agents or oxidative stress [62–64]. The best-studied enzyme of the PARP family, PARP-1, is characterized as an abundant nuclear protein that binds SSBs and double strand DNA breaks (DSBs) [64]. PARP-1 is involved in DNA repair, recombination, and genomic stability [65, 66] of extensive DNA breakage that is triggered by massive oxidative or nitrosative stress [67–69]. The overactivation of PARP depletes the cellular stores of NAD+ and ATP [69], leading to a severely compromised cellular energetic state that inhibits apoptotic cell death and results in necrotic cell death, followed by the activation of inflammatory processes that augment tumor development.
In all related cases, the initiation potential of oxidants seems to contribute to carcinogenesis by inducing DNA base changes in certain oncogenes and tumor suppressor genes [70–72]. It is shown that the hydroxyl radical (HO·) is able to activate certain oncogenes, such as K-ras and C-Raf-1. Activation proceeds through the induction of point mutations in CpG base pairs and gene deletions [72]. Point mutations in CpG dinucleotides are frequently found in certain tumor suppressor genes, such as p53 and ras, leading to inactivation [60]. Furthermore, HO· attacks cells containing mutant or absent p53, resulting in failure to arrest in G1, and reducing cell capacity to repair damaged DNA [60, 72]. This increase in replication errors can initiate additional oncogene activation and tumor suppressor gene inactivation, ultimately contributing to malignancy. Free radical-induced cytotoxicity may also initiate carcinogenesis by depleting the normal cell population and promoting the clonal expansion of more resistant cells, thus increasing the probability of mutation [60].
The roles of GSH and taurine in cancer prevention or promotion
Considering the mechanisms of tumor induction, it is reasonable to assume that the trans-sulfuration pathway is necessary to maintain intracellular redox homeostasis. Alterations in this pathway increase the rate of base mispairing due to base oxidation, abasic sites, error-prone recombination events, alterations in gene expression, and chronic inflammatory processes, thus inducing tumor progression (Fig. 3B). Another mechanism that could contribute to tumor induction is the depletion of functional proteins by RS, resulting in an increase in oxidized protein level (Fig. 3B). Once formed, oxidized proteins need to be recycled by the ubiquitin system, increasing the amino acid requirement for protein synthesis, especially methionine and cysteine [4]. Assuming that cysteine levels are maintained by the breakdown of GSH [11, 73], there is a shift in the redox balance following the induction of oxidative stress, such that the ratio of GSH to GSSG (the oxidized form of GSH) is altered in favor of GSSG [11, 73].
In addition to its function as a cysteine repository, GSH is the most prevalent non-protein thiol in mammalian cells and the major low molecular-weight peptide present in eukaryotic cells [73]. GSH acts as a reducing agent, is involved in the metabolism of xenobiotics, is a free-radical scavenger, aids in cell-cycle regulation and microtubular-related mechanisms, regulates Ca2+ homeostasis, regulates protein function and gene expression via thiol-disulfide exchange reactions, modulates lymphocyte function and immune responses, and participates in the mitochondrial mechanisms that link the opening of the permeability transition pore complex to the activation of cell death [74]. Since GSH has such a wide range of functions, alterations in GSH levels are associated with numerous human diseases, including cancer, neurodegeneration, acquired immune deficiency syndrome (AIDS), aging, cystic fibrosis, liver diseases, heart attack, stroke, seizure, diabetes, sickle cell anemia, and kwashiorkor [74, 75].
Glutathione also appears to be very important for tumor development and growth. Hirono [76] was the first to show that ascites tumor cells, which are highly resistant to alkylating agents, have higher GSH levels than non-tumor cells. Different types of tumor, including melanoma, bladder carcinoma, lung cancer, colon cancer, and breast tumors that are multidrug and radiation resistant, are also found to have a high GSH content [77–81]. Considering the homeostatic redox buffer function of GSH and its role in inactivating some carcinogens [82] and protecting cells against DNA-damaging free radicals and lipid peroxidation [83], it is plausible that tumor cells may need more GSH for their survival than other cell types. This is supported by results showing that levels of GSH and enzymes related to the GSH synthesis pathway (Fig. 1) fluctuate over a wide range of concentrations (up to 40-fold), regardless of treatment status or primary tumor origin [84]. One of the best known enzymes in GSH metabolism, glutathione-S-transferase (GSTs; EC 2.5.1.18) is shown to be associated with multidrug resistance of tumor cells [85]. GSTs belong to the family of phase II detoxification enzymes that catalyze GSH (S-glutathionylation) conjugation with different chemotherapeutic compounds to form mercapturates [86], which are easily excreted in urine, thus diminishing the therapeutic effects of antitumor drugs. In addition, GST overexpression can increase susceptibility to carcinogenesis and inflammatory disease [86], which is associated with tumor progression. High GSH concentrations in cancer cells, induction of GSH in murine melanocytes following c-H-ras oncogene-induced transformation, or impairment of metastatic spread by GSH depletion have also been described, further highlighting the importance of GSH pathways in tumor development and growth [87, 88]. Under metastatic conditions, high levels of GSH can support (i) a rapid cell cycle, (ii) an elevated rate of DNA synthesis, and (iii) a block in apoptosis [74]. Thus, GSH can be characterized as a double edged sword, protecting non-tumor cells against oxidative stress induced by metabolism or exogenous compounds and at the same time, protecting tumor cells from apoptosis and chemotherapeutic treatments, thus furthering tumor development and metastasis. A recent study by Uthus et al. [89] demonstrated that the Ames dwarf mouse has significantly increased hepatic GSH levels because of an absence of growth hormone (GH). The lack of GH in dwarf mice results in higher overall levels of tissue glutathione S-transferase and GSH [90], enhancing cellular detoxification and providing resistance against toxic/oxidative stress [89]. It is also reported that Ames dwarf mice live 50–64% longer than their wild type littermates [90] because of the delayed occurrence and reduced incidence of fatal neoplasic disease. These data support the hypothesis that heightened antioxidative defenses may prolong life span [90]. Thus, the Ames dwarf mouse may be more resistant to oxidative stress because it has a relatively large pool of GSH as a result of altered levels of GSH metabolites and other sulfur-containing molecules.
In its turn, taurine is a conditionally essential non-proteinogenic amino acid that is required for many aspects of mammalian metabolism [91]. Taurine is required for the development and survival of mammalian cells, being the most abundant single amino acid in leukocytes (20–50 mM) [92–95]. Taurine also protects the lung from ozone, bleomycin, nitrogen dioxide, and amiodarone induced injury [96–99]. In addition, taurine can protect cells from oxidant-induced injury by forming taurine chloramine (Tau-Cl). Tau-Cl synthesis occurs through a reaction between taurine and plasma hypochlorous acid (HOCl) [100], resulting in the inactivation of this strong oxidant and cytotoxic agent. HOCl is the final product of the reaction between H2O2 and chloride (Cl-) ion that is catalyzed by myeloperoxidase (MPO; EC 1.11.1.7). HOCl is also found in equilibrium with molecular chlorine (Cl2), and responsible for damaging lipids, proteins, and DNA [101, 102]. High levels of HOCl production by polymorphic variants of MPO are associated with chronic inflammation, which increases the risk of leukemia, lung cancer, and laryngeal cancer [101, 102]. Chronic inflammation induced by elevated levels of HOCl is also observed in atherosclerotic tissue [103], a common condition in hyperhomocyst(e)inuric patients. HOCl can halogenate/oxidize pyrimidine and purine bases of DNA, generating the highly mutagenic 5-chloro-2′-deoxycytidine (5-CldC) [102]. 8-Chlorinated products of 2′-deoxyadenosine and 2′-deoxyguanosine, as well as semistable or unstable chloramines (RNHCl and RR’NCl) [101] are also generated from the reaction with HOCl [102], Unstable nucleoside chloramines, such as thymidine chloramine, are shown to initiate DNA single and double strand breaks through nitrogen-centered radicals and to transfer chlorine atoms to other nucleosides [101]. While uracil concentrations are normally low in plasma, the dissolution of dead cells by necrosis, which is induced by PARP enzyme overexpression [64] and hyperhomocyst(e)inuria, liberates nucleobases and nucleosides into the extracellular environment [102]. This phenomenon increases the uracil content in inflamed tissue fluid (which is as high as 600 μM) to almost 1000-fold higher than the plasma level [102] and provides MPO with ample substrate for halogenation. Thus, all of the necessary factors for pyrimidine halogenation are provided by the inflammatory milieu [101, 102]. Furthermore, because inflammation also causes cells to proliferate, it sets the stage for thymine analogs like halogenated nucleobases to be misincorporated into newly synthesized DNA, which induces mutations [102]. The inflammation-mediated chlorination of cytosine residues in DNA may also account for several DNA alterations observed in human tumors, including alterations in methylation patterns [104].
Under normal conditions, taurine controls cell and tissue levels of Cl− and HOCl by reacting with these molecules to generate Tau-Cl [91]. In addition, Tau-Cl downregulates the immunologic response by reducing the production of proinflammatory mediators like nitric oxide, tumor necrosis factor-K, prostaglandin E2, and monocyte chemotactic protein-1, which blocks the cascade effect of chronic inflammation that leads to tumor progression [91].
Is the methionine dependency of tumor cells an adaptation?
Considering that chronic inflammation and mutagenesis are important factors for tumor development, and that metabolic deficiencies in the trans-sulfuration pathway are directly correlated with chronic inflammation (especially deficiencies that result in cellular homocyst(e)ine accumulation), it is likely that the elevated production of homocyst(e)ine by methionine-dependent cancer cells is an adaptation that has allowed tumor cells to survive and colonize a constantly changing, biological environment. In this sense, atherosclerosis and cancer share many common mechanisms related to disease development and progression, both being a consequence of complex interactions between genetic and environmental factors [105]. Recently, a number of reports have suggested that both atherosclerosis and cancer develop from a clonal proliferation of altered cells at the sight of local tissue injury, inflammation, and genomic instability [105–107]. Atherosclerosis may begin when an injury or infection transforms a single arterial smooth-muscle progenitor of a proliferative clone, which resembles carcinogenesis [108, 109]. Atherosclerotic plaque and malignant tumor formation are associated with (i) cell proliferation regulatory pathways, (ii) alterations in adhesion molecule expression, (iii) alterations in protease expression, (iv) altered ligand-growth factor interactions, (v) altered nuclear transcription factor expression, and (vi) the production of angiogenesis modulators [105]. The infiltration of macrophages into the cellular mass [110] is another common characteristic of atherosclerotic lesions and tumors. Since Rudolf Virchow first demonstrated that the inflammatory process influences atherosclerosis and tumor development in the 19th century [111], a growing body of evidence has shown that macrophages play an important role in initiating and promoting both pathologies [110, 111]. For example, microbial and viral infections induce a chronic immunologic response that results in a persistent inflammatory process that could result in tumor development. The induction of a chronic inflammatory process by the tumor itself or its leukocyte-infiltrate may also accelerate cancer growth and metastasis [112, 113]. In both cases, the combined effects of ROS, cytokines, chemokines, as well as angiogenic factors produced by tumor-associated macrophages (TAMs) and other inflammatory cells explains the abnormal growth of healthy cells [110, 114]. Although the molecular mechanisms of macrophage-inducing tumor and atherosclerosis development remain poorly understood, it is well established that once a cellular mass becomes infiltrated by macrophages, the ability of tumor and atherosclerotic tissue to survive the immune response increases exponentially. If we consider that methionine-dependent tumors produce large amounts of homocyst(e)ine from methionine (Fig. 4), and hyperhomocyst(e)inuric individuals are at increased risk of developing atherosclerosis, it is plausible that homocysteine is one of the signals required to recruit macrophages to the site of tumor and atherosclerotic cells (Fig. 4). It is possible that tumor cells induce a local hyperhomocyst(e)inuria that recruits a subpopulation of macrophages (Fig. 4). Once established, the macrophages trigger a series of biochemical events that result in the increased synthesis of proinflammatory molecules, RS, halogenated pyrimidines, angiogenic and growth factors, and create an optimal cellular microenvironment (Fig. 4). This may lead to the selection and stimulation of cells with the ability to evade the immune system and colonize other tissues. Evidence for a role of homocyst(e)ine in the recruitment of macrophages and induction of inflammatory processes further supports this idea [115]. Further studies will be needed to analyze the effect of local hyperhomocyst(e)inuria that has resulted from trans-sulfuration deficiencies on the development of optimal microenvironments for tumor and atherosclerosis development.

Schematic model of a cancer microenvironment induced by chronic inflammation and local hyperhomocyst(e)inuria. assuming that methionine-dependent tumor cells produce high levels of homocysteine and generate a localized hyperhomocyst(e)inuria, macrophages are recruited by the release of reactive oxygen species (ROS). The combination of ROS, homocyst(e)ine and parp overexpression, induces tumor cell necrosis, releasing high amounts of pyrimidines into the tumor mass. the pyrimidine molecules are converted into halogenated pyrimidines by macrophage myeloperoxidase and HOCL. the halogenated pyrimidines are strong mutagenic substances that (i) increase the genetic diversity of tumor cells and help to select the best-adapted cells, and (ii) convert some macrophages into tumor-associated macrophages (TAMs). once established with the tumor cells, TAMs initiate angiogenic and growth factor synthesis that contributes to the expansion and maintenance of a cancer microenvironment. in addition, the induction of a constant, chronic inflammatory response by macrophages results in the liberation of proinflammatory molecules that induce necrosis and increase the velocity of tumor cell division
Concluding remarks
The trans-sulfuration pathway is an important biochemical mechanism that links methionine metabolism to the production of redox-controlling molecules. While genetic and biochemical knowledge about the trans-sulfuration pathway has been accumulated in the past few years, further research is required to assess the physiological importance of this pathway in eukaryotic cells. Deficiencies in certain components of the trans-sulfuration pathway are linked to the development of various pathologies in humans. For example, excessive homocyst(e)ine production, observed in particular trans-sulfuration pathway deficiencies, may result in a chronic inflammatory process that leads to atherosclerosis development. It is also observed that the excessive production of homocyst(e)ine may activate macrophages and induce proinflammatory molecule synthesis that is associated with the development and selection of tumor cells, and the establishment of optimal biochemical and environmental conditions for metastasis. In this sense, a study analyzing the importance of the trans-sulfuration pathway in tumor and atherosclerotic development, the recruitment of macrophages for active growing cellular masses, and the creation of microenvironments that select active growing cells is urgently required in order to effectively design new chemotherapeutical molecules or protocols.
References
Cellarier E, Durando X, Vasson MP, Farges MC, Demiden A, Maurizis JC, Madelmont JC, Chollet P (2003) Methionine dependency and cancer treatment. Cancer Treat Rev 29:489–499
Kokkinakis DM (2006) Methionine-stress: a pleiotropic approach in enhancing the efficacy of chemotherapy. Cancer Lett 233:195–207
Finkelstein JD, Martin JJ (2000) Homocysteine. Int J Biochem Cell Biol 32:385–389
Stipanuk MH (2004) Sulfur amino acid metabolism: pathways for production and removal of homocysteine and cysteine. Annu Rev Nutr 24:539–577
Nobori T, Szinai I, Amox D, Parker B, Olopade OI, Buchhagen DL, Carson DA (1993) Methylthioadenosine phosphorylase deficiency in human non-small cell lung cancers. Cancer Res 53:1098–1101
Traweek ST, Riscoe MK, Ferro AJ, Braziel RM, Magenis RE, Fitchen JH (1988) Methylthioadenosine phosphorylase deficiency in acute leukemia: pathologic, cytogenetic, and clinical features. Blood 71:1568–1573
Nobori T, Karras JG, Della Ragione F, Waltz TA, Chen PP, Carson DA (1991) Absence of methylthioadenosine phosphorylase in human gliomas. Cancer Res 51:3193–3197
Fitchen JH, Riscoe MK, Dana BW, Lawrence HJ, Ferro AJ (1986) Methylthioadenosine phosphorylase deficiency in human leukemias and solid tumors. Cancer Res 46:5409–5412
Mato JM, Corrales FJ, Lu SC, Avila MA (2002) S-Adenosylmethionine: a control switch that regulates liver function. FASEB J 16:15–26
Ulrey CL, Liu L, Andrews LG, Tollefsbol TO (2005) The impact of metabolism on DNA methylation. Hum Mol Genet 14:R139-R147
Lu SC (1999) Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J 13:1169–1183
Tarver H, Schmidt CLA (1939) The conversion of methionine to cystine: experiments with radioactive sulfur. J Biol Chem 130:67–80
Beatty PW, Reed DJ (1980) Involvement of the cystathionine pathway in the biosynthesis of glutathione by isolated rat hepatocytes. Arch Biochem Biophys 204:80–87
Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R (2006) S-adenosylmethionine stabilizes cystathionine beta-synthase and modulates redox capacity. Proc Natl Acad Sci USA 103:6489–6494
Cooper AJL (1983) Biochemistry of sulfur-containing amino acids. Annu Rev Biochem 52:187–222
Mosharov E, Cranford MR, Banerjee R (2000) The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 39:13005–13011
Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR, Banerjee R (2004) Perturbations in homocysteine-linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol 287:R39-R46
Martinov MV, Vitvitsky VM, Mosharov EV, Banerjee R, Ataullakhanov FI (2000) A substrate switch: a new mode of regulation in the methionine metabolic pathway. J Theor Biol 204:521–532
Neuhauser-Berthold M, Kuhfus A, Bassler KH (1988) Utilization of glutathione disulfide as cysteine source during long-term parenteral nutrition in the growing rat. Metabolism 37:796–801
Lu SC, Huang HY (1994) Comparison of sulfur amino acid utilization for GSH synthesis between HepG2 cells and cultured rat hepatocytes. Biochem Pharmacol 47:859–869
Guo H, Lishko V, Herrera H, Groce A, Kubota T, Hoffman RM (1993) Therapeutic tumor-specific cell-cycle block induced by MET starvation in vivo, Cancer Res 53:5676–5679
Kokkinakis DM, Schold SC, Hori H, Nobori T (1997) Effect of long-term depletion of plasma MET on the growth and survival of human brain tumor xenografts in athymic mice. Nutr Cancer 29:195–204
Halpern BC, Clark BR, Hardy DN, Halpern RM, Smith RA (1974) The effect of replacement of methionine by homocystine on survival of malignant and normal adult mammalian cells in culture. Proc Natl Acad Sci USA 71:1133–1136
Breillout F, Antoine E, Poupon MF (1990) Methionine dependency of malignant tumors: a possible approach for therapy. J Natl Cancer Inst 82:1628–1632
Stern PH, Hoffman RM (1986) Enhanced in vitro selective toxicity of chemotherapeutic agents for human cancer cells based on a metabolic defect. J Natl Cancer Inst 76:629–639
Guo HY, Herrera H, Groce A, Hoffman RM (1993) Expression of the biochemical defect of methionine dependence in fresh patient tumors in primary histoculture. Cancer Res 53:2479–2483
Judde JG, Ellis M, Frost P (1989) Biochemical analysis of the role of transmethylation in the MET dependence of tumor cells. Cancer Res 29:4859–4865
Palmer JL, Abeles RH (1979) Mechanism of action of S-adenosylhomocysteinase. J Biol Chem 254:1217–1226
Kloor D, Osswald H (2004) S-Adenosylhomocysteine hydrolase as a target for intracellular adenosine action. Trends Pharmacol Sci 25:294–297
Fujioka M, Takata Y (1981) S-Adenosylhomocysteine hydrolase from rat liver. Purification and some properties J Biol Chem 256:1631–1635
Hu Y, Komoto J, Huang Y, Gomi T, Ogawa H, Takata Y, Fujioka M, Takusagawa F (1999) Crystal structure of S-adenosylhomocysteine hydrolase from rat liver. Biochemistry 38:8323–8333
Turner MA, Yuan CS, Borchardt RT, Hershfield MS, Smith GD, Howell PL (1998) Structure determination of selenomethionyl S-adenosylhomocysteine hydrolase using data at a single wavelength. Nat Struct Biol 5:369–376
Kloor D, Fuchs S, Petroktistis F, Delabar U, Muhlbauer B, Quast U, Osswald H (1998) Effects of ions on adenosine binding and enzyme activity of purified S-adenosylhomocysteine hydrolase from bovine kidney. Biochem Pharmacol 56:1493–1496
Mohandas T, Sparkes RS, Suh EJ, Hershfield MS (1984) Regional localization of the human genes for S-adenosylhomocysteine hydrolase (cen----q131) and adenosine deaminase (q131----qter) on chromosome 20. Hum Genet 66:292–295
Baric I, Fumic K, Glenn B, Cuk M, Schulze A, Finkelstein JD, James SJ, Mejaski-Bosnjak V, Pazanin L, Pogribny IP, Rados M, Sarnavka V, Scukanec-Spoljar M, Allen RH, Stabler S, Uzelac L, Vugrek O, Wagner C, Zeisel S, Mudd SH (2004) S-adenosylhomocysteine hydrolase deficiency in a human: a genetic disorder of methionine metabolism. Proc Natl Acad Sci USA 101:4234–4239
Gellekink H, den Heijer M, Kluijtmans LA, Blom HJ (2004) Effect of genetic variation in the human S-adenosylhomocysteine hydrolase gene on total homocysteine concentrations and risk of recurrent venous thrombosis. Eur J Hum Genet 12:942–948
Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M (1999) Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat 13:362–375
Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R (1998) Evidence for hememediated redox regulation of human cystathionine β-synthase activity. J Biol Chem 273:25179–25184
Meier M, Janosik M, Kery V, Kraus JP, Burkhard P (2001) Structure of human cystathionine β-synthase: a unique pyridoxal 5’-phosphate-dependent heme protein. EMBO J 20:3910–3916
Oliveriusova J, Kery V, Maclean KN, Kraus JP (2002) Deletion mutagenesis of human cystathionine β-synthase: impact on activity, oligomeric status, and S-adenosylmethionine regulation. J Biol Chem 277:48386–48394
Shan X, Dunbrack RL, Christopher SA, Kruger WD (2001) Mutations in the regulatory domain of cystathionine β-synthase can functionally suppress patient-derived mutations in cis. Hum Mol Genet 10:635–643
Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP (2001) Regulation of human cystathionine β-synthase by S-adenosyl-l-methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry 40:10625–10633
Zou CG, Banerjee R (2003) Tumor necrosis factor-α-induced targeted proteolysis of cystathionine β-synthase modulates redox homeostasis. J Biol Chem 278:16802–16808
Yang G, Cao K, Wu L, Wang R (2004) Cystathionine γ-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J Biol Chem 279:49199–49205
Seshadri S, Beiser A, Selhub J, Jacques PF, Rosenberg IH, D’Agostino RB, Wilson PW, Wolf PA (2002) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346:476–483
Weiss N, Heydrick S, Zhang Y -Y, Bierl C, Cap A, Loscalzo J (2002) Cellular redox state and endothelial dysfunction in mildly hyperhomocysteinemic cystathionine β-synthase-deficient mice. Arterioscler Thromb Vasc Biol 22:34–41
Wu LL, Wu JT (2002) Hyperhomocysteinemia is a risk factor for cancer and a new potential tumor marker. Clin Chim Acta 322:21–28
Xu D, Neville R, Finkel T (2000) Homocysteine accelerates endothelial cell senescence. FEBS Lett 470:20–24
Ishii I, Akahoshi N, Yu X–N, Kobayashi Y, Namekata K, Komaki G, Kimura H (2004) Murine cystathionine γ-lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 381:113–123
Messerschmidt A, Worbs M, Steegborn C, Wahl MC, Huber R, Laber B, Clausen T (2003) Determinants of enzymatic specificity in the cys-met-metabolism PLP-dependent enzymes family: crystal structure of cystathionine γ-lyase from yeast and intrafamiliar structure comparison. Biol Chem 384:373–386
Wang J, Hegele RA (2003) Genomic basis of cystathioninuria (MIM 219500) revealed by multiple mutations in cystathionine gamma-lyase (CTH). Hum Genet 112:404–408
Wong LT, Hardwick DF, Applegarth DA, Davidson AG (1979) Review of metabolic screening program of children’s hospital, Vancouver, British Columbia. 1971–1977 Clin Biochem 12:167–172
Lemieux B, Auray-Blais C, Giguere R, Shapcott D, Scriver CR (1988) Newborn urine screening experience with over one million infants in the Quebec Network of Genetic Medicine. J Inherit Metab Dis 11:45–55
Letavayová L, Marková E, Hermanská K, Vlčková V, Vlasákova D, Chovaneca M, Brozmanova J (2006) Relative contribution of homologous recombination and non-homologous end-joining to DNA double-strand break repair after oxidative stress in Saccharomyces cerevisiae. DNA Repair 5:602–610
Demple B, Harrison L (1994) Repair of oxidative damage to DNA: enzymology and biology. Annu Rev Biochem 63:915–948
Laval J (1996) Role of DNA repair enzymes in the cellular resistance to oxidative stress. Pathol Biol 44:14–24
Wallace SS (1998) Enzymatic processing of radiation-induced free radical damage in DNA. Radiat Res 150:S60-S79
Bjelland S, Seeberg E (2003) Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res Fundam Mol Mech Mut 531:37–80
Slupphaug G, Kavli B, Krokan HE (2003) The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res 531:231–251
Valko M, Izakovic M, Mazur M, Rhodes CJ, Telser J (2004) Role of oxygen radicals in DNA damage and cancer incidence. Mol Cell Biochem 266:37–56
Cerutti PA, Trump BF (1991) Inflammation and oxidative stress in carcinogenesis. Cancer Cells 3:1–7
Tasatargil A, Sadan G, Karasu E (2006) Homocysteine-induced changes in vascular reactivity of guinea-pig pulmonary arteries: role of the oxidative stress and poly (ADP-ribose) polymerase activation. Pulm Pharmacol Ther (in press)
Martinet W, Knaapen MWM, De Meyer GRY, Herman AG, Kockx MM (2002) Elevated levels of oxidative DNA damage and DNA repair enzymes in human atherosclerotic plaques. Circulation 106:927–932
Virág L, Szabó C (2002) The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev 54:375–429
Bouchard VJ, Rouleau M, Poirier GG (2003) PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol 31:446–454
Burkle A (2001) Physiology and pathophysiology of poly(ADP-ribosyl)ation. BioEssays 23:795–806
Stenerlow B, Karlsson KH, Cooper B, Rydberg B (2003) Measurement of prompt DNA double-strand breaks in mammalian cells without including heat-labile sites: results for cells deficient in nonhomologous end joining. Radiat Res 159:502–510
Sutherland BM, Bennett PV, Sidorkina O, Laval J (2000) Clustered DNA damages induced in isolated DNA and in human cells by low doses of ionizing radiation. Proc Natl Acad Sci USA 97:103–108
Audebert M, Salles B, Calsou P (2004) Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem 279:55117–55126
Guyton KZ, Kensler TW (1993) Oxidative mechanisms in carcinogenesis. Br Med Bull 49:523–544
Cerda S, Weitzman SA (1997) Influence of oxygen radical injury on DNA methylation. Mut Res Rev Mutat Res 386:141–152
Jackson JH (1994) Potential molecular mechanisms of oxidant-induced carcinogenesis. Environ Health Perspect 102:155–158
Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND (2004) Glutathione metabolism and its implications for health. J Nutr 134:489–492
Estrela JM, Ortega A, Obrador E (2006) Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci 43:1–39
Townsend DM, Tew KD, Tapiero H (2003) The importance of glutathione in human disease. Biomed Pharmacother 57:145–155
Hirono I (1961) Mechanism of natural and acquired resistance to methyl-bis-(beta-chlorethyl)-amine N- oxide in ascites tumors. Gann 52:39–48
Carretero J, Obrador E, Anasagasti MJ, Martin JJ, Vidal-Vanaclocha F, Estrela JM (1999) Growth–associated changes in glutathione content correlate with liver metastatic activity of B16 melanoma cells. Clin Exp Metastasis 17:567–574
Pendyala L, Velagapudi S, Toth K, Zdanowicz J, Glaves D, Slocum H, Perez R, Huben R, Creaven PJ, Raghavan D (1997) Translational studies of glutathione in bladder cancer cell lines and human specimens. Clin Cancer Res 3:793–798
Honda T, Coppola S, Ghibelli L, Cho SH, Kagawa S, Spurgers KB, Brisbay SM, Roth JA, Meyn RE, Fang B, McDonnell TJ (2004) GSH depletion enhances adenoviral bax-induced apoptosis in lung cancer cells. Cancer Gene Ther 11:249–255
Berger SJ, Gosky D, Zborowska E, Willson JK, Berger NA (1994) Sensitive enzymatic cycling assay for glutathione: measurements of glutathione content and its modulation by buthionine sulfoximine in vivo and in vitro in human colon cancer. Cancer Res 54:4077–4083
Perry RR, Mazetta JA, Levin M, Barranco SC (1993) Glutathione levels and variability in breast tumors and normal tissue. Cancer 72:783–787
Jakobisiak M, Lasek W, Golab J (2003) Natural mechanisms protecting against cancer. Immunol Lett 90:103–122
Sadani GR, Nadkarni GD (1996) Role of tissue antioxidant defence in thyroid cancers. Cancer Lett 109:231–235
Schadendorf D, Jurgovsky K, Kohlmus CM, Czarnetzki BM (1995) Glutathione and related enzymes in tumor progression and metastases of human melanoma. J Invest Dermatol 105:109–112
Townsend DM, Tew KD (2003) The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 22:7369–7375
Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88
Revesz L, Edgren MR, Wainson AA (1994) Selective toxicity of buthionine sulfoximine (BSO) to melanoma cells in vitro and in vivo. Int J Radiat Oncol Biol Phys 29:403–406
Thrall BD, Meadows GG (1991) Induction of glutathione content in murine melanocytes after transformation with c-H-ras oncogene. Carcinogenesis 12:1319–1323
Uthus EO, Brown-Borg HM (2006) Methionine flux to transsulfuration is enhanced in the long living Ames dwarf mouse. Mech Ageing Dev 127:444–450
Brown-Borg HM, Rakoczy SG, Uthus EO (2005) Growth hormone alters methionine and glutathione metabolism in Ames dwarf mice. Mech Ageing Dev 126:389–398
Park E, Park SY, Wang C, Xu J, LaFauci G, Schuller-Levis G (2002) Cloning of murine cysteine sulphonic acid decarboxylase and its mRNA expression in murine tissues. Biochim Biophys Acta 1574:403–406
Sturman JA (1993) Taurine in development. Physiol Rev 73:119–147
Hayes KC, Carey RE, Schmidt SY (1975) Retinal degeneration associated with taurine deficiency in the cat. Science 188:949–951
Fukuda K, Hirai Y, Yoshida H, Hakajima T, Usii T (1982) Free amino acid content of lymphocytes and granulocytes compared. Clin Chem 28:1758–1761
Green T, Fellman JH, Eicher AL, Pratt KJ (1991) Antioxidant role and subcellular location of hypotaurine and taurine in human neutrophils. Biochim Biophys Acta 1073:91–97
Schuller-Levis G, Gordon RE, Park E, Pendino KJ, Laskin D (1995) Taurine protects rat bronchioles from acute ozone-induced lung inflammation and hyperplasia. Exp Lung Res 21:877–888
Wang QJ, Giri SN, Hyde DM, Nakashima JM (1989) Effects of taurine on bleomycin-induced lung fibrosis in hamsters. Proc Soc Exp Biol Med 190:330–338
Gordon RE, Shaked AA, Solano DF (1986) Taurine protects hamster bronchioles from acute NO2-induced alterations. A histologic, ultrastructural, and freeze-fracture study Am J Pathol 125:585–600
Wang Q, Hollinger MA, Giri SN (1992) Attenuation of amiodarone-induced lung fibrosis and phospholipidosis in hamsters by taurine and/or niacin treatment. J Pharmacol Exp Ther 262:127–132
Schuller-Levis GB, Park E (2003) Taurine: new implications for an old amino acid. FEMS Microbiol Lett 226:195–202
Kawai Y, Morinaga H, Kondo H, Miyoshi N, Nakamura Y, Uchida K, Osawa T (2004) Endogenous formation of novel halogenated 2′-deoxycitidine. J Biol Chem 279:51241–51249
Henderson JP, Byun J, Takeshita J, Heinecke JW (2003) Phagocytes produce 5-chlorouracil and 5-bromouracil, two mutagenic products of myeloperoxidase, in human inflammatory tissues. J Biol Chem 278:23522–23528
McMillen TS, Heinecke JW, LeBoeuf RC (2005) Expression of human myeloperoxidase by macrophages promotes atherosclerosis in mice. Circulation 111:2798–2804
Valinluck V, Liu P, Kang JI, Burdzy A, Sowers LC (2005) 5-Halogenated pyrimidine lesions within a CpG sequence context mimic 5-methylcytosine by enhancing the binding of the methyl-CpG-binding domain of methyl-CpG-binding protein 2 (MeCP2). Nucleic Acids Res 33:3057–3064
Li JJ, Gao R-L (2005) Should atherosclerosis be considered a cancer of the vascular wall? Med Hypotheses 64:694–698
Ross JS, Stagliano NE, Donovan MJ, Breitbart RE, Ginsburg GS (2001) Atherosclerosis and cancer: common molecular pathway of disease development and progression. Ann N Y Acad Sci 947:271–292
Ross JS, Stagliano NE, Donovan MJ, Breitbart RE, Ginsburg GS (2001) Atherosclerosis: a cancer of blood vessels. Am J Clin Pathol 116:S97–S107
Shackelford RE, Kaufmann WK, Paules RS (2000) Oxidative stress and cell cycle checkpoint function. Free Rad Biol Med 28:1387–1404
Bartsch H (2000) Studies on biomarkers in cancer etiology and prevention: a summary and challenge of 20 years of interdisciplinary research. Mutat Res 462:V255–V279
Lamagna C, Aurrand-Lions M, Imhof BA (2006) Dual role of macrophages in tumor growth and angiogenesis. J Leukoc Biol 80:705−713
Heidland A, Klassen A, Rutkowski P, Bahner U (2006) The contribution of Rudolf Virchow to the concept of inflammation: what is still of importance? J Nephrol 19:S102-S109
Schwartsburd PM (2003) Chronic inflammation as inductor of procancer microenvironment: pathogenesis of dysregulated feedback control. Cancer Metastasis Rev 22:95–102
Negus RP, Stamp GW, Hadley J, Balkwill FR (1997) Quantitative assessment of the leukocyte infiltrate in ovarian cancer and its relationship to the expression of C-C chemokines. Am J Pathol 150:1723–1734
Smith GR, Missailidis S (2004) Cancer, inflammation and the AT1 and AT2 receptors. J Inflamm 1:3
Sharma P, Senthilkumar RD, Brahmachari V, Sundaramoorthy E, Mahajan A, Sharma A, Sengupta S (2006) Mining literature for a comprehensive pathway analysis: A case study for retrieval of homocysteine related genes for genetic and epigenetic studies. Lipids Health Dis 5:1
Acknowledgments
We thank Dr. Kátia Gonçalves dos Santos for critically reading the manuscript. This work was supported by research grants from Fundação de Amparo a Pesquisa do Estado do Rio Grande do Sul (FAPERGS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Rosado, J.O., Salvador, M. & Bonatto, D. Importance of the trans-sulfuration pathway in cancer prevention and promotion. Mol Cell Biochem 301, 1–12 (2007). https://doi.org/10.1007/s11010-006-9389-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-006-9389-y