
Abstract
Mutter’s pseudoproline dipeptides and Sheppard’s Hmb derivatives are powerful tools for enhancing synthetic efficiency in Fmoc SPPS. They work by exploiting the natural propensity of N-alkyl amino acids to disrupt the formation of the secondary structures during peptide assembly. Their use results in better and more predictable acylation and deprotection kinetics, enhanced reaction rates, and improved yields of crude products. However, these approaches have certain limitations: pseudoproline dipeptides can only be used for sequences containing serine or threonine, and the coupling of the amino acid following the Hmb residue can be extremely difficult. To alleviate some of these shortcomings, we have prepared a range of Fmoc-Aaa-(Dmb)Gly-OH dipeptides and tested their efficacy in the synthesis of a number of challenging hydrophobic peptides. We also compared the efficiency of N-Dmb against N-Hmb backbone protection in preventing aspartimide formation in the Fmoc SPPS of peptides containing the Asp-Gly sequence.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The propensity of certain peptide sequences to form β-sheets and other secondary structures on the solid phase is well recognized as the cause of failure sequences during peptide assembly (Kent 1988). In t-butoxycarbonyl-based (Boc) solid phase peptide synthesis (SPPS), the strategy of using neat trifluoroacetic acid (TFA) for Boc group removal in combination with in situ neutralization during coupling appears to be extremely effective at overcoming this problem (Schnölzer et al. 1992). Unfortunately, until now no equivalent generic strategy for fluorenylmethoxycarbonyl-based (Fmoc) SPPS has been developed.
Many researchers have attempted to overcome aggregation in Fmoc SPPS by improving the solvation of the peptide chain through the use of either chaotropic reagents, (Stewart and Klis 1990), complex solvents mixtures (Zhang et al. 1994), and polar resins (Bayer 1991). However, none of these approaches has found widespread acceptance, owing either to their lack of general applicability, partial effectiveness or incompatibility with standard protocols. A more systemic approach, and one which addresses the underlying causes of aggregation, namely secondary structure formation, involves the use of building blocks incorporating reversible backbone amide protection. It has been long known from guest–host studies on highly structured amino-acid homopolymers that insertion of secondary amino acids, such as proline (Toniolo et al. 1981) or N-alkyl amino acid residues (Narita et al. 1984), at an appropriate location within such a sequence can disrupt β-sheets and helices. The effect can last for up to six residues from the point of insertion of the secondary amino acid (Bedford et al. 1992) and has been shown to operate in solution, the solid state, and most importantly on a resin during peptide assembly (Milton et al. 1990). This phenomenon provided the principle behind the development of Sheppard’s 2-hydroxyl-4-methoxybenzyl (Hmb) (Johnson et al. 1993) and Mutter’s pseudoproline derivatives (Haack and Mutter 1992) shown in Fig. 1. These building blocks temporarily introduce a structure-disrupting secondary amino acid residue into a peptide sequence. Their use have been shown to result in better and more predictable acylation and deprotection kinetics, enhanced reaction rates, and improved yields of crude products (Packman et al. 1994; Hyde et al. 1994; Simmonds 1996; White et al. 2004; Abedini and Raleigh 2005). On completion of the assembly, treatment with TFA cleaves off backbone protection, regenerating the natural amino acid.

Structure breaking building blocks—pseudoproline proline and Hmb-/Dmb-amide protection
These approaches do have, however, certain limitations: pseudoproline dipeptides can only be used for sequences containing serine or threonine, and the coupling of the amino acid following the Hmb residue can be extremely difficult (Sampson et al. 1999). From our perspective, the concept of incorporating the secondary amino acid residue using a pre-formed dipeptide, as in the case of pseudoproline dipeptides, is particularly attractive since it avoids difficulties with forming tertiary amide bonds on the solid support. Moreover, such dipeptides can be coupled using the standard activation chemistries employed on automated peptide synthesizers without modification of existing protocols. Therefore, in order to extend the scope of structure-breaking dipeptides beyond peptides containing Ser and Thr residues, we have prepared a range of backbone amide protected dipeptides of the general formula Fmoc-Aaa-(Dmb)Gly-OH. 2,4-dimethoxybenzyl (Dmb) was selected for protection of the backbone as it has a long pedigree in peptide synthesis (Weygand et al. 1966), is removed with TFA, and cannot form lactones as is the case with Hmb (Nicolas et al. 1997). Whilst, our approach is only appropriate for introduction of (Dmb)Gly and cannot be applied to chiral amino acids, as the corresponding dipeptides would be at risk of epimerization during coupling, we nevertheless believe such dipeptides would prove valuable tools for the synthesis of hydrophobic and amyloidogenic peptides since glycine occurs frequently in such sequences.
To demonstrate the efficacy of our Dmb-dipeptides derivatives, we report here the Fmoc synthesis of the amyloidogenic Neurotoxin prion peptide (PrP) 106–126 (Jobling et al. 1999) and the highly hydrophobic transmembrane domain of rat bradykinin b2 receptor (Oliveira et al. 1997). Both peptides reportedly could not be made by Fmoc SPPS. We also compared the efficiency of N-Dmb against N-Hmb backbone protection in preventing aspartimide formation in the Fmoc SPPS of peptides containing the Asp-Gly sequence.
Materials and Methods
All amino acid derivatives and peptide synthesis supports were obtained from Merck Biosciences AG (Laeufelfingen, Switzerland). All solvents used were of analytical grade or peptide synthesis grade and were used without further purification.
Syntheses on the ABi 433 peptide synthesiser were carried out using standard FastMoc protocols as supplied by the manufacturer, with the exception that Fmoc removal was effected by treating the resin three times for 2 min with 20% piperidine. Coupling reactions were performed using Fmoc amino acids (10 eq.) activated with HBTU/HOBt (1:1, 10 eq.) and DIPEA (20 eq.). For peptides prepared on the Protein Technologies Symphony, Fmoc-amino acids (10 eq.) were pre-dissolved in DMF and activated with HCTU (10 eq.) and NMM (20 eq.). Fmoc groups were removed by two treatment with 20% piperidine in DMF (2 min, 10 min). Manual syntheses were performed as described in the results and discussion section. The sidechains of trifunctional amino acids were protected as follows: Arg(Pbf), Asn(Trt), Asp(OtBu), Gln(Trt), Glu(OtBu), Lys(Boc), Ser(tBu), Thr(tBu), Trp(Boc),Tyr(tBu).
Unless otherwise stated, cleavage of peptides from the resin with concomitant side-chain deprotection was effected by treating peptidyl resins with TFA/water/triisopropylsilane (TIS) (95:2.5:2.5 v/v) for 3 h. Following removal of the TFA and scavengers by evaporation under vacuum, the peptides were precipitated by the addition of diethyl ether or diisopropyl ether. The peptides were isolated by filtration and washed three times with clean ether.
Results and Discussion
The synthesis of amyloidogenic and transmembrane peptides represents a significant challenge for peptide chemists, owing to their propensity to aggregate during peptide assembly. In order to evaluate the utility of our Dmb-dipeptides in expediting the synthesis of such peptides, we selected sequences from the literature representative of each class of difficult peptide (Table 1, peptides 1 and 2).
Neurotoxin prion peptide (PrP) 106–126 (Table 1, peptide 1) comprising residues 106–126 of human PrP has been found to be highly amyloidogenic and toxic to neurons (Tagliavini et al. 1993). The SPPS of PrP (106–126) has been described by Jobling et al. (1999). In their hands, the synthesis of this peptide using standard Fmoc-protected amino acids was unsuccessful, with chain extension terminating at Ala117. Incorporation of (Hmb)Gly residues at positions 114 and 119 resulted in some improvement in synthetic efficiency and afforded the full length peptide in a yield of only 7.3%. However, the target peptide could only be obtained in good yield and purity by using Boc-protected amino acids with in situ neutralization coupling protocols. The difficulties in the synthesis of PrP (106–126) were ascribed to the inherent propensity of this peptide to aggregate, particularly in the region of the AGAAAAGA sequence.
In view of the problems experienced by Jobling et al., we decided to prepare this peptide using three Dmb dipeptides at the positions marked in bold in Table 1. This approach would introduce structure-breaking (Dmb)Gly residues before Ala120 and Ala117, regions of the sequence which were found by Jobling et al. to be particularly problematic, and would therefore maximize the chances of a successful synthesis. The peptide was prepared using Fmoc-Gly-Wang resin (0.1 mmol, 0.71 mmol/g) on an ABi 433A peptide synthesizer using standard FastMoc protocols with 30 min coupling times. The Dmb dipeptides were introduced manually using 3-fold excesses of reagents instead of the standard 10-fold excess which was used for all the other amino acids. Treatment of the resin with TFA/water/TIS afforded the crude PrP in excellent purity, and MS analysis of the product showed no evidence of deletion or truncated peptides (Figs. 2a, 3a). Two further syntheses of PrP were also carried out, one using Ala-(Dmb)Gly and Gly-Gly(Dmb) dipeptides at residues 118,119 and 123,124, respectively, and another using a single Ala-(Dmb)Gly at residues 113, 114. In both cases these products were of significantly lesser quality indicating the need for the use of three Dmb dipeptides.

HPLC profiles of crude PrP prepared using a three Dmb-dipeptides and b standard Fmoc-amino acid derivatives. HPLC conditions: column: Merck Chromolith; buffer A: 0.1% TFA aq., buffer B: MeCN/water/TFA (90:10:0.1); gradient: 0.2 min at 5% B then to 100% B in 18 min; flow rate: 3 ml/min; detection: 220 nm

ES-MS spectra of crude PrP prepared using a three Dmb-dipeptides and b standard Fmoc-amino acid derivatives. ESI: M+H+expected 1913.2, found 1913.2
The synthesis of PrP (106–126) was then repeated using standard Fmoc-amino acid building blocks, and PrP was cleaved as described previously. In contrast to the synthesis of Jobling et al. where none of the target peptide was obtained, our crude product did contain the desired peptide as the major component. However, characterization of this material by LC-MS indicated that the product contained also significant amounts of the des-Asn108 and des-(Lys106, Asn108) deletion peptides together with smaller amounts of other peptides missing residues from the N-terminal sequence (Figs. 2b, 3b). Furthermore, the desLys, Asn-PrP (106–126) coeluted with the product making the mixture inseparable.
The preparation of the N-cysteinyl analog of the transmembrane domain of rat bradykinin b2 receptor (TM-33) (Table 1, peptide 2) was first described by Oliveira et al. (1997). It was chosen as a model to study the problems associated with the synthesis and purification of hydrophobic transmembrane sequences. The best results (12% yield) were obtained using Boc chemistry with 20% DMSO in NMP as reaction solvent. The synthesis using optimized Fmoc SPPS methods, however, was unsuccessful and did not result in the formation of any detectable product, despite the application of Fmoc-(Hmb) derivatives of Ala and Leu to prevent aggregation.
For our investigations we decided to study the synthesis of a version of TM-34 which omits the N-terminal Cys residue, to avoid complications due to dimerization via disulfide bond formation. TM-33 was first assembled on Fmoc-Asp(OtBu)-Wang resin using a Protein Technologies Symphony peptide synthesizer. Couplings were carried out for 1 h using 10-fold excesses of standard Fmoc-amino acid building blocks activated with HCTU/NMM. Global deprotection and cleavage from the resin was carried out as previously described. The highly insoluble product was dissolved in neat TFA, which was diluted with MeCN/water, and analyzed by HPLC (Fig. 4) and MALDI-TOF. The product was found to be highly heterogeneous and not to contain any major component, confirming the findings of Oliveira et al. (1997).

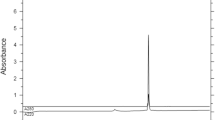
HPLC profile of crude TM-34 prepared using standard Fmoc-amino acid derivatives. HPLC conditions as given in Fig. 2
The synthesis was then repeated using the same conditions with a combination of pseudoproline dipeptides and Dmb-dipeptides. Ile-Thr/Ala-Ser and Ala-Gly were introduced using Fmoc-Ile-Thr(ψMe,Mepro)-OH/Fmoc-Ala-Ser(ψMe,Mepro)-OH and Fmoc-Ala-(Dmb)Gly-OH, respectively. Treatment with TFA as previously described afforded the desired peptide in excellent purity as determined by HPLC and MALDI-TOF (Fig. 5).

a HPLC profiles and b MALDI-TOF spectrum of crude TM-34 prepared using pseudoproline and Dmb-dipeptides. HPLC conditions: column: Zorbax SB-C18; buffer A: 0.1% TFA aq., buffer B: 0.1% TFA in MeCN; gradient: 0.5–100% B in 35 min; flow rate: 0.8 ml/min; detection: 215 nm. MALDI-TOF: M+Na+ expected 3487.9, found 3487.6
Aspartimide Formation
Temporary backbone-amide protection of glycine with the TFA-labile Hmb group has been shown by Quibell et al. (1994) to totally eliminate aspartimide formation during the Fmoc SPPS of peptides containing Asp-Gly. This approach works by blocking the amide nitrogen, thereby preventing it from attacking the carbonyl of the β-ester (Fig. 6).

Prevention of aspartimide formation by backbone-amide protection
Introduction of (Hmb)Gly was originally effected using Fmoc-(FmocHmb)Gly-OH. However, the difficulties associated with acylation of the glycine secondary amine on the solid phase led Mergler et al. (2003) to develop the dipeptide derivative Fmoc-Asp(OtBu)-(Hmb)Gly-OH in which the tertiary amide bond is preformed. In our hands, Fmoc-Asp(OtBu)-(Hmb)Gly-OH was found to be extremely effective at preventing aspartimide formation but very slow to couple, which we attributed to lactone formation between the unprotected phenolic hydroxyl and carboxyl group. We were, therefore, very interested to compare the ease of introduction and efficacy of this compound against that of our Fmoc-Asp(OtBu)-(Dmb)Gly-OH derivative. This compound has also been described by Zahariev et al. (2007). We selected the known aspartimide prone sequence H-Val-Lys-Asp-Gly-Tyr-Leu-NH2 (Table 1, peptide 3) as a model for our studies. Peptidyl resins 4–7 (Table 2) were prepared manually on Rink Amide MBHA resin with 30 min couplings of 2-fold excess of amino acid derivatives activated with TBTU/DIPEA (1:2). Fmoc removal was achieved by two treatments for 15 min with 20% piperidine in NMP. Completeness of coupling reactions was monitored using TNBS and chloranil tests, where appropriate. Peptidyl resin 5 was prepared using Fmoc-(Dmb)Gly-OH since we were interested to observe how easily H-(Dmb)Gly could be acylated on the solid phase, as of Zahariev et al. (2007) had reported that this could be accomplished without difficultly.
Notable differences were observed in the ease of assembly of our model peptide, depending on which derivatives were used for the incorporation of the Asp-Gly sequence. As anticipated, and in contrast to the findings of Zahariev et al. (2007), acylation of the resin-bound secondary amine was indeed found to be difficult in the synthesis of peptide 5, requiring double coupling to achieve complete addition of the Asp residue. Incorporation of Fmoc-Asp(OtBu)-(Hmb)Gly-OH into peptide 6 took 1.5 h, compared to 30 min for Fmoc-Asp(OtBu)-(Dmb)Gly-OH in peptide 7.
The crude products were cleaved and side-chain deprotected by treatment of the peptidyl resins with TFA, as previously described, and analyzed and characterized by LC-MS. HPLC profiles of the crude peptides are shown in Fig. 7. As expected, the product from resin 4, which was prepared using standard Fmoc-amino acid derivatives, contained a considerable quantity of aspartimide by-products. Resins 5 & 7 made utilizing Dmb-backbone protection gave products of excellent quality, with negligible aspartimide formation. In the case of resin 6, which was prepared using Hmb-backbone protection, little aspartimide formation was observed, but the product did contain a significant quantity of a yet unidentified material (Table 3).

HPLC profiles of crude peptide 3 obtained from peptidyl resins a 4, b 5, c 6, d 7. HPLC conditions: column: Nucleosil 300 C-18; buffer A: 0.1% TFA aq., buffer B: 0.1% TFA in MeCN; gradient: 5–95% B in 30 min; flow rate: 1 ml/min; detection: 215 nm
Conclusions
Dmb-dipeptides are powerful additions to the peptide chemist’s armoury for overcoming aggregation during Fmoc SPPS. They complement Mutter’s pseudoproline dipeptides by expanding the possible sites for incorporation of structure-breaking secondary amino acids beyond serine and threonine to include glycine as well. This capability should greatly enhance the range of difficult and complex peptides that can be made utilizing this strategy. It is anticipated that these derivatives will prove particularly useful in the synthesis of difficult, hydrophobic transmembrane peptides as glycine residues are commonly found in such sequences.
The dipeptide Fmoc-Asp(OtBu)-(Dmb)Gly-OH has been shown to be highly effective at preventing aspartimide formation during the Fmoc SPPS of Asp-Gly-containing peptides. Its efficacy coupled with its ease of introduction under standard coupling methods should make it the reagent of choice for overcoming this side reaction.
References
Abedini A, Raleigh DP (2005) Incorporation of Pseudoproline derivatives allows the facile synthesis of human IAPP, a highly amyloidogenic and aggregation-prone polypeptide. Org Lett 7:693–696
Bayer E (1991) Towards the chemical synthesis of proteins. Angew Chem Int Ed Engl 30:113–129
Bedford J, Carolyn BH, Johnson T, Jun W, Quibell M, Robert CS (1992) Amino acid structure and difficult sequences in solid phase peptide synthesis. Int J Pept Protein Res 40:300–307
Haack T, Mutter M (1992) Serine derived oxazolidines as secondary structure disrupting, solubilizing building blocks in peptide synthesis. Tetrahedron Lett 33:1589–1592
Hyde C, Johnson T, Owen D, Quibell M, Sheppard RC (1994) Some difficult sequences made easy—a study of interchain association in solid-phase peptide-synthesis. Int J Pept Protein Res 43:431–440
Jobling MF, Barrow CJ, White AR, Masters CL, Collins SJ, Cappai R (1999) The synthesis and spectroscopic analysis of the neurotoxic prion peptide 106–126: comparative use of manual Boc and Fmoc chemistry. Lett Pept Sci 6:129–134
Johnson T, Quibell M, Owen D, Sheppard RC (1993) A reversible protecting group for the amide bond in peptides: use in the synthesis of difficult sequences. J Chem Soc Chem Commun 369–372
Kent SBH (1988) Chemical Synthesis of Peptides and Proteins. Annu Rev Biochem 57:957–989
Mergler M, Dick F, Sax B, Weiler P, Vorherr T (2003) The aspartimide problem in Fmoc-based SPPS: part 1. J Pept Sci 9:36–46
Milton RCD, Milton SCF, Adams PA (1990) Prediction of difficult sequences in solid-phase peptide synthesis. J Am Chem Soc 112:6039–6046
Narita M, Fukunaga T, Wakabayashi A, Ishikawa K, Nakano H (1984) Syntheses and properties of tertiary peptide-bond-containing polypeptides. Int J Pept Protein Res 23:306–314
Nicolas E, Pujades M, Bacardit J, Giralt E, Albericio F (1997) A new approach to Hmb-backbone protection of peptides: synthesis and reactivity of N-alpha-Fmoc-N-alpha-(Hmb) amino acids. Tetrahedron Lett 38:2317–2320
Oliveira E, Miranda A, Albericio F, Andreu D, Paiva ACM, Nakaie CR, Tominaga M (1997) Comparative evaluation of the synthesis and purification of transmembrane peptide fragments–rat bradykinin receptor fragment 64–97 as model. J Pept Res 49:300–307
Packman LC, Quibell M, Johnson T (1994) Roles of electrospray mass-spectrometry, counterion distribution monitoring and N-(2-Hydroxy-4-Methoxybenzyl) backbone protection in peptide-synthesis. Pept Res 7:125–131
Quibell M, Owen D, Packman LC, Johnson T (1994) Suppression of piperidine-mediated side product formation for Asp(OtBu)-containing peptides by the use of N-(2-Hydroxy-4-Methoxybenzyl) (Hmb) backbone amide protection. J Chem Soc Chem Commun 2343–2344
Sampson WR, Patsiouras H, Ede NJ (1999) The synthesis of ‘Difficult’ peptides using 2-Hydroxy-4-methoxybenzyl or pseudoproline amino acid building blocks: a comparative study. J Pept Sci 5:403–409
Schnölzer M, Alewood P, Jones A, Alewood D, Kent SBH (1992) In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences. Int J Pept Protein Res 40:180–193
Simmonds RG (1996) Use of the Hmb backbone-protecting group in the synthesis of difficult sequences. Int J Pept Protein Res 47:36–41
Stewart JM, Klis WA (1990) Polystyrene-based solid phase peptide synthesis: the state-of-the-art. In: Epton E (ed) Innovations and perspectives in solid phase synthesis: peptides, polypeptides and oligonucleotides. SPCC (UK) Ltd, Birmingham, pp 1–9
Tagliavini F, Prelli F, Verga L et al (1993) Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc Natl Acad Sci USA 90:9678–9682
Toniolo C, Bonora GM, Mutter M, Pillaib VNR (1981) linear oligopeptides, 77a) the effect of the insertion of a proline residue on the solid-state conformation of host peptides. Makromol Chem 182:1997–2005
Weygand F, Steglich W, Bjarnason J, Akhtar R, Khan M (1966) Leicht abspaltbare schutzgruppen für säureamidfunktionen 1 Mitteilung. Tetrahedron Lett 7:3483–3487
White PD, Keyte J, Bailey K, Bloomberg G (2004) Expediting the Fmoc solid phase synthesis of long peptides through the application of dimethyloxazolidine dipeptides. J Pept Sci 10:18–26
Zahariev S, Guarnaccia C, Pongor CI, Quaroni L, Cemazar M, Pongor S (2007) Application of peptoid methodology for synthesis of “Difficult” peptides free of aspartimide and related products. In: Rolka K, Rekowski P, Silberring J (eds) Peptides 2006, Proceedings of the European Peptide Symposium. Kennes International, Geneva, pp 84–85
Zhang L, Goldammer C, Henkel B, Zuehl F, Panhaus G, Jung G, Bayer E (1994) “Magic Mixture”, a powerful solvent system for solid-phase synthesis of “difficult sequences”. In: Epton E (ed) Innovation and perspectives in solid phase synthesis: peptides, proteins and nucleic acids—biological and biomedical applications, 3rd International Symposium. Mayflower Worldwide Ltd., Birmingham, UK, pp 711–716
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cardona, V., Eberle, I., Barthélémy, S. et al. Application of Dmb-Dipeptides in the Fmoc SPPS of Difficult and Aspartimide-Prone Sequences. Int J Pept Res Ther 14, 285–292 (2008). https://doi.org/10.1007/s10989-008-9154-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10989-008-9154-z