
Abstract
Gaps in our understanding of muscle mechanics demonstrate that the current model is incomplete. Increasingly, it appears that a role for titin in active muscle contraction might help to fill these gaps. While such a role for titin is increasingly accepted, the underlying molecular mechanisms remain unclear. The goals of this paper are to review recent studies demonstrating Ca2+-dependent interactions between N2A titin and actin in vitro, to explore theoretical predictions of muscle behavior based on this interaction, and to review experimental data related to the predictions. In a recent study, we demonstrated that Ca2+ increases the association constant between N2A titin and F-actin; that Ca2+ increases rupture forces between N2A titin and F-actin; and that Ca2+ and N2A titin reduce sliding velocity of F-actin and reconstituted thin filaments in motility assays. Preliminary data support a role for Ig83, but other Ig domains in the N2A region may also be involved. Two mechanical consequences are inescapable if N2A titin binds to thin filaments in active muscle sarcomeres: (1) the length of titin’s freely extensible I-band should decrease upon muscle activation; and (2) binding between N2A titin and thin filaments should increase titin stiffness in active muscle. Experimental observations demonstrate that these properties characterize wild type muscles, but not muscles from mdm mice with a small deletion in N2A titin, including part of Ig83. Given the new in vitro evidence for Ca2+-dependent binding between N2A titin and actin, it is time for skepticism to give way to further investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fundamental gaps in our understanding of muscle contraction are illustrated by recent studies. At the level of muscle mechanics, the currently accepted sliding-filament–swinging cross-bridge paradigm of muscle contraction, typically modeled as cross-bridge motors in series with elastic elements (Fenn 1923; Hill 1922; Huxley 1957, 1973), fails to predict several important muscle properties. These include the persistent increase in muscle force after active stretch (Lindstedt and Nishikawa 2017; Nishikawa et al. 2018a), elastic recoil of muscles during rapid unloading (Sugi et al. 2000), and the dependence of muscle mechanical behavior on the phase of activation in stretch–shortening cycles (Ahn et al. 2003). At the level of biomechanics, models based on the current paradigm fail to yield accurate predictions of in vivo muscle forces in humans and animals during natural movements (Dick et al. 2017; Lee et al. 2013).
Early studies naturally attributed muscle properties to the cross bridges. Examples include instantaneous elasticity (Huxley and Simmons 1971a) and energy storage during active stretch (Lombardi et al. 1995). More recently, it has been recognized that other structures, including thin and thick filaments as well as titin, contribute to muscle compliance (Wakabayashi et al. 1994) and energy storage (Linari et al. 2003). In fact, recent estimates suggest that cross bridges account for only a small fraction (up to ~ 12%) of energy storage in muscles during active stretch, with only 2% from cross bridge elasticity (Linari et al. 2003).
The current sliding filament–swinging cross-bridge paradigm was developed decades before the discovery of titin in the late 1970’s (Maruyama et al. 1976; Wang et al. 1979), so it is understandable that a role for titin in active muscle contraction was overlooked initially. Increasingly, it appears that a role for titin in active muscle contraction might help to fill gaps in the current paradigm (Lindstedt and Nishikawa 2017; Linke 2018). Acceptance of the idea that titin contributes to active muscle force production has led to a profusion of new proposals for mechanisms (Linke 2018). Several new mechanisms range from direct effects on titin mechanical properties, such as equilibrium length and stiffness (Leonard and Herzog 2010; Nishikawa 2016; Nishikawa et al. 2012) and Ig domain unfolding (Rivas-Pardo et al. 2016), to indirect effects on cross-bridge force production (Mijailovich et al. 2019), potentially mediated via thick (Irving 2017) and thin filament structural changes, as well as other biochemical interactions and signaling pathways (Pfuhl and Gautel 2012).
The goals of this paper are to review recent studies demonstrating Ca2+-dependent interactions between N2A titin and actin in vitro, to explore theoretical predictions of muscle mechanical behavior based on this interaction, and to review experimental data related to the predictions. The analysis suggests that titin may contribute importantly to muscle force production, and that future progress in muscle research will likely require better integration of bottom-up studies on molecular mechanisms with top-down studies on muscle mechanics and in vivo muscle function.
Titin function: from passive scaffold to regulation of active muscle force
Early work on titin interactions with myosin (Murayama et al. 1989) and actin (Maruyama et al. 1987) demonstrated that titin forms a scaffold connecting the thick and thin filaments in sarcomeres of striated muscles (Gautel and Djinovic-Carugo 2016; Linke et al. 1999). In the Z-disc, titin Z-repeats bind to F-actin (Trombitas and Granzier 1997), alpha-actinin (Linke et al. 1997), and telethonin (Gregorio et al. 1998, 1999). In the A-band, repeating titin FnIII domains bind to the tail portion of myosin (Houmeida et al. 1995), as well as to MyBP-C (Labeit et al. 1992) and myomesin (Obermann et al. 1996). Numerous other proteins were ultimately found to interact with the giant titin protein from M-line to Z-disk in muscle sarcomeres (Gautel and Djinovic-Carugo 2016; Gregorio et al. 1999; Sanger and Sanger 2001).
In the I-band, the mechanical properties of tandem Ig and PEVK domains of titin (Fig. 1) produce the passive tension associated with myofibrils, muscle fibers (Linke et al. 1998a, b) and intact muscles (Brynnel et al. 2018). Titin passive tension is thought to maintain sarcomere structure in both longitudinal (Horowits et al. 1986; Horowits and Podolsky 1987) and radial directions (Irving et al. 2011; Konhilas et al. 2002; Li et al. 2016).

Reproduced with permission from Nishikawa (2016). Copyright 2016, The Company of Biologists Ltd
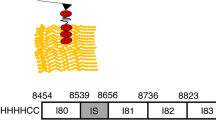
Layout of titin and other proteins in skeletal muscle sarcomeres. Each titin molecule is bound to the thin filaments (blue) in the Z-disc (blue triangles on left side of half-sarcomere) and to the thick filaments (purple) in the A-band. The N2A segment (red) is located between the proximal tandem Ig segments (orange) and the PEVK segment (green). Ig, immunoglobulin
The possibility that titin’s role in muscle sarcomeres might extend beyond passive tension and maintenance of sarcomere structural integrity to regulation of active muscle contraction was considered even in the earliest studies on titin (Maruyama et al. 1976). Only recently, however, has this idea become more widely accepted (see e.g., (Linke 2018); (Freundt and Linke 2018). A recent study demonstrates that titin contributes not only to passive force, but also to active force of skeletal muscles (Li et al. 2018). In this study, a transgenic mouse was developed in which a specific proteolytic cleavage site from tobacco etch virus was inserted into the distal I-band region of titin near the edge of the A-band. When titin was cleaved in fiber bundles from homozygous transgenic mice, both passive and active force of muscle fibers decreased by ~ 50%.
Searching for Ca2+-dependent titin–actin interactions: absence of evidence is not evidence of absence
The idea that titin could play a role in regulating active force production in striated muscles motivated the search for Ca2+-dependent interactions with other proteins (Maruyama et al. 1976), as the cytoplasmic calcium transient is the major signal that triggers muscle force production via activation of thin filaments in skeletal muscle. The search naturally focused on titin interactions with F-actin and thin filaments in the I-band, since A-band titin runs along the thick filaments and is relatively inextensible (Trinick 1996). Early investigations of Ca2+-dependent titin–actin interactions focused on binding of native titin (Maruyama et al. 1987; Soteriou et al. 1993) or titin fragments (Kellermayer and Granzier 1996) to F-actin and thin filaments. Until recently (Dutta et al. 2018), the only experimental evidence supporting the existence of Ca2+-dependent interactions between titin and thin filaments in the I-band of skeletal muscle came from Kellermayer and Granzier (1996), who demonstrated, using in vitro motility and binding assays, that the large T2 fragment of titin (comprising residues from the N2-line to M-line) interacts with actin filaments and reconstituted thin filaments at pCa < 6.0. They showed that the motility of actin filaments on heavy meromyosin was reduced by the T2 fragment in the presence of Ca2+.
The availability of the titin gene sequence from humans (Labeit and Kolmerer 1995) and rats (Jin 1995, 2000; Li et al. 1995) provided the opportunity to clone and purify titin constructs. A large number of studies (Bianco et al. 2007; Kulke et al. 2001; Linke et al. 1997, 2002; Nagy et al. 2004; Yamasaki et al. 2001), employing a variety of methods including co-sedimentation and other binding assays, in vitro motility assays, and optical tweezers, examined Ca2+-dependence of titin–actin interactions using titin constructs based on gene sequences from human soleus (GenBank X90569) and cardiac (GenBank X90568) titin. Focusing mostly on PEVK residues, these studies found that constructs based on soleus titin (GenBank X90569, residues 5618–7791) exhibited only Ca2+-independent interactions with actin (Linke et al. 2002; Nagy et al. 2004; Yamasaki et al. 2001), whereas interactions between F-actin and constructs based on cardiac titin (GenBank X90568) were inhibited by the calcium binding protein S100A1 when Ca2+ was present (Yamasaki et al. 2001).
Given the large number of previous studies investigating interactions between titin constructs and F-actin, it is surprising that none included a sequence of 115 amino acids (residues 5508–5617 from X90569) within the Ig83 domain of N2A titin (Dutta et al. 2018). Only Linke et al. (1997) examined interactions between N2A titin (X90569, residues 5010–5507, Ig80-IS-Ig81-Ig82) and actin. Using immunofluorescence microscopy and co-sedimentation experiments, they found no interactions between N2A titin and F-actin or reconstituted thin filaments outside the Z-disc in the presence or absence of Ca2+. Notably however, the terminal Ig83 domain from the N2A region was not included in the constructs they tested (Linke et al. 1997). The missing 115 amino acid sequence includes the last 53 C-terminal amino acids of Ig83, which are also deleted in the ‘muscular dystrophy with myositis’ (mdm) mutation in mice (Garvey et al. 2002).
New evidence for Ca2+-dependent interactions between N2A titin and thin filaments
In a recent study (Dutta et al. 2018), we hypothesized that the N2A region of titin, overlooked in previous studies, could be responsible for Ca2+-dependent interactions of the titin T2 fragment with actin and reconstituted thin filaments observed previously by Kellermayer and Granzier (1996). We used co-sedimentation, in vitro motility assays (IVM), and dynamic force spectroscopy (DFS) to characterize interactions between a recombinantly expressed N2A titin construct (Ig80-IS-Ig81-Ig82-Ig83) and F-actin or reconstituted thin filaments in the presence and absence and Ca2+.
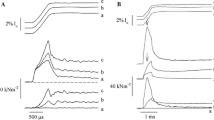
In this study, we demonstrated that the recombinantly expressed N2A construct co-sediments with F-actin and that Ca2+ increases the association constant between the N2A construct and F-actin (Fig. 2). Actin binding depends on the concentration of both N2A and Ca2+, with a significant increase in binding observed with higher N2A concentrations and higher [Ca2+] (ANCOVA, p < 0.0003), suggesting that Ca2+ stabilizes interactions between N2A and F-actin (Dutta et al. 2018). This hypothesis was further supported by single molecule force spectroscopy experiments demonstrating increased rupture forces (Fig. 3, t test, p < 0.001) and decreased off-rate (Dutta et al. 2018) in the presence of Ca2+ (pCa = 4.0). Finally, we demonstrated that the N2A constructs reduce sliding velocity of F-actin and reconstituted thin filaments (Ca2+-regulated due to the presence of troponin and tropomyosin) using in vitro motility (IVM) assays. Statistically significant decreases in velocity were measured for both types of filaments at pCa ≤ 5 (Fig. 4, ANOVA, p < 0.0001; (Dutta et al. 2018), a concentration of Ca2+ that is physiologically relevant with respect to activation of skeletal muscle. Taken together, these results demonstrate the potential for N2A titin to interact with thin filaments in muscle sarcomeres, and suggest that the strength of that interaction is regulated by Ca2+ concentration. The high rupture forces (~ 100 pN; Fig. 3) measured between the N2A construct and F-actin at pCa = 4 suggest that, if binding occurs in vivo, it could potentially account for all of the energy stored in muscle during active stretch (Linari et al. 2003).

Reproduced with permission from Dutta et al. (2018)
Ca2+ increases co-sedimentation of N2A constructs with actin filaments. a Example SDS-PAGE gel of 3 µM N2A bound to actin at pCa = 4 (squares) and pCa = 10 (diamonds). The gel was cropped to highlight the region containing the 3 µM N2A samples. b Plot of average band intensities of bound N2A versus N2A concentration at pCa 10 and pCa 4 (N = 3 per group). ANOVA demonstrates significant effects of [N2A] (p < 0.0001), [Ca2+] (p = 0.0003), and their interaction (p = 0.0125) on N2A–F-actin binding as estimated by densitometry. P pellet, S supernatant

Reproduced with permission from Dutta et al. (2018)
Ca2+ increases rupture forces of interacting N2A and F-actin molecules. Overlay of force-extension curves from single molecule force spectroscopy in the a absence (pCa = 10) and b presence (pCa = 4) of Ca2+. Histograms of N2A–F-actin rupture forces and most probable rupture force at a loading rate of 8000 pN/s in the c absence of Ca2+ (mean rupture force of 70 ± 40 pN) and d presence of Ca2+ (mean rupture force of 100 ± 40 pN). Two tailed t-test demonstrates that Ca2+ significantly increases the strength of N2A–F-actin interactions (p < 0.001)

Reproduced with permission from Dutta et al. (2018)
Ca2+ decreases in vitro motility of F-actin and reconstituted thin filaments. a The N2A construct (triangles) decreases motility of F-actin in the presence of Ca2+ compared to BSA controls (circles). ANOVA demonstrates that the velocity of F-actin decreased significantly in the presence of N2A versus BSA (p < 0.0001) and also decreased with increasing [Ca2+] (p < 0.0001). b In vitro motility of reconstituted thin filaments decreases in the presence of N2A at pCa = 4.0 and 5.0. ANOVA demonstrates significant effects of N2A versus BSA (p < 0.0001), [Ca2+] (p < 0.0001), and their interaction (p < 0.0001) on the velocity of regulated filaments. Asterisks indicate a significant decrease in filament velocity in the presence of the N2A construct at pCa 4 and 5 (Tukey’s HSD, p < 0.05). Fitting the data using the Hill cooperativity equation demonstrated that pCa50 was unchanged in the presence of N2A, but N2A increased cooperativity (Hill coefficients for BSA control = 0.88, N2A = 1.564) and decreased Vmax (N2A = 3.13) compared to BSA control (4.09)
While stable binding might require additional Ig domains, preliminary force spectroscopy data from constructs of individual Ig domains suggest that Ig83 contains amino acids required for Ca2+-dependent binding to F-actin. Preliminary actin binding assay experiments provide further support for this model. This would explain why titin–actin interactions were observed in our studies (Dutta et al. 2018), but were absent in Linke et al. (1997), as the constructs used in that study included most of the N2A region, but did not include Ig83. These data are also consistent with the loss of function observed in skeletal muscles from mdm mice, with an 83 amino acid deletion in I-band titin, 53 amino acids of which are deleted from Ig83 and the remaining 30 from the proximal PEVK region (Garvey et al. 2002). These observations suggest that the mdm mouse is a good model for testing hypotheses about the contribution of Ca2+-dependent titin–actin interactions to muscle mechanics.
Future studies are needed to provide additional insights into mechanism(s) for Ca2+ modulation of N2A–thin filament binding. However, a few potential mechanisms are consistent with current data. While the possibility exists that Ca2+ stabilizes the N2A–actin interaction through non-specific binding along the interface, it is more common for modulated biological events to be regulated through specific interactions. We therefore favor a mechanism in which Ca2+ binds to specific sites on the N2A construct. Indeed, previous studies (Tatsumi et al. 2001) demonstrate that ~ 12 molecules of calcium bind to a 400 kDa fragment of titin that includes the N2A region. The N2 line in muscle sarcomeres (Yarom and Meiri 1971), from which N2A gets its name, was originally discovered based on staining with pyroantimonate which precipitates in the presence of calcium.
Our working hypothesis is that Ig domains in the N2A region contain calcium binding sites and that Ca2+ stabilizes those conformation(s) capable of binding to actin filaments. While our preliminary data suggest that Ig83 is the primary binding site, further experiments are underway to determine the specific domains involved in N2A–actin binding. If the binding site for Ca2+ is located in the Ig83 domain, it will not be a classical EF-hand domain. The EF-hand motif is composed of α-helices but titin Ig domains are composed of seven β-strands in a two-layered sandwich (von Castelmur et al. 2008). We predict that binding is likely to involve clusters of glutamates within Ig83. However, a high-resolution structure will be necessary in order to determine the exact nature of the calcium binding site(s). Ongoing work is focused on determining which Ig domains in the N2A region bind to actin, quantifying their stability, and determining the specific residues and mechanisms that are involved. The binding kinetics of calcium to N2A have not yet been determined, but binding would need to be in the micromolar range to be consistent with the calcium-based effects that were measured in previous studies (Dutta et al. 2018). Once these details are known, it would be interesting to investigate whether Ca2+-binding to N2A titin is strain-dependent.
Additional predictions are that the stability of the Ig domains composing the N2A region should vary as a function of [Ca2+], and that domains involved in binding would show the largest change in stability in the presence versus absence of Ca2+. This could be investigated using chemical stability or differential scanning calorimetry to determine how Ca2+ affects the stability of various regions of N2A. As characterization of the N2A region is completed, the mechanism of Ca2+-based stabilization of the N2A–actin interaction will become increasingly clear.
Reported evidence against titin–actin interactions in muscle sarcomeres?
Two recent studies have argued that titin–actin interactions do not occur in muscle sarcomeres. One recent study reported evidence against titin–actin interactions in single myofibrils. Cornachione et al. (2016) used gelsolin to extract actin from single myofibrils to test for effects of titin–actin interactions on muscle mechanical properties. They found only a small difference in stretching force before and after actin removal, using fluorescent phalloidin to visualize actin extraction. While their images showed reduced actin in the A-band, Z-line actin appeared unaffected and moreover (Zhukarev et al. 1997), demonstrated that phalloidin does not stain actin in the I-bands of sarcomeres despite the known presence of actin in this region. They further suggested that nebulin might block binding of phalloidin to actin in the I-band (Zhukarev et al. 1997). Because phalloidin does not bind to actin in the I-band, the methods employed by Cornachione et al. (2016) fail to indicate whether gelsolin had in fact removed actin from the I-band where N2A titin is expected to bind, and the experiments therefore cannot refute the existence of titin–actin interactions in this region as the authors originally claimed (Nishikawa 2016).
A second study (DuVall et al. 2017) used the F146 antibody, which binds to titin in the distal PEVK segment near the edge of the A-band, to investigate segmental elongation of titin during passive and active stretch. They found evidence for an effect of Ca2+ but no apparent evidence for interactions with actin on the extension of titin segments proximal and distal to the antibody label during stretch. Critically, however, the F146 antibody binds to titin at a location that is far removed from the N2A region. Therefore, the proximal titin segment between the Z-disc and the F146 antibody includes not only N2A but most of the PEVK segment, making it relatively insensitive to changes in elongation of the proximal tandem Ig region per se.
A more definitive test of whether N2A titin binds to actin in skeletal muscle sarcomeres requires use of antibodies that bind closer to the N2A region, either in the N2A region itself or between N2A and the Z-line. If N2A titin binds to actin upon Ca2+-activation as suggested (Dutta et al. 2018), then elongation of tandem Ig domains proximal to N2A (Fig. 5a, red), which normally occurs during passive stretch (Linke et al. 1998b) should be prevented during active stretch (Fig. 5b, orange) of sarcomeres.

Predicted segmental elongation of titin in passively and actively stretched sarcomeres. a Passive stretch: the proximal segment is expected to stretch more than the distal segment due to its lower stiffness. b Active stretch: N2A binding to actin is expected to prevent elongation of the proximal segment, so that only the distal segment elongates. c Elongation of titin segments in passively and actively stretched sarcomeres labeled with N2A antibodies. During passive stretch, the distal PEVK segment (green) extends earlier and faster than expected and the proximal Ig domains (purple) extend less than expected. A similar pattern was observed during active stretch (blue = distal segment, red = proximal segment). d Reduced elongation of the proximal segment in sarcomeres of one myofibril activated at longer SL. In all sarcomeres of this myofibril, the proximal segment remained at a constant length during active stretch up to lengths of ~ 4.0 μm as expected. Inset shows average extension of proximal Ig domains (black) in sarcomeres activated at longer initial length
We tracked the motion of N2A titin in myofibrils from wild type mouse extensor digitorum longus (EDL) by labeling titin with a primary N2A antibody and fluorescent secondary antibodies. The primary N2A antibody binds to titin at Ig80-81 (Freiburg et al. 2000), and extension of titin segments proximal and distal to the fluorescent label was determined from the movement of the labeled N2A relative to myofibril and sarcomere extension. Extension was measured during passive (no Ca2+) and active stretch from real time video images. We expected to observe more lengthening of the proximal segment (Fig. 5a, red) than the distal segment (Fig. 5a, blue) in passive myofibrils due to low force straightening of the tandem Ig region. We also expected to observe that the proximal segment (Fig. 5b, orange) would fail to elongate in active myofibrils if N2A titin binds to actin.
In all passive myofibrils, the distal PEVK segment elongated at shorter sarcomere lengths than expected based on previous work (Linke et al. 1998a). During both passive (n = 7 myofibrils, 49 sarcomeres, Fig. 5c, green line) and active stretch (n = 10 myofibrils, 55 sarcomeres, Fig. 5c, blue line), the distal segments of most sarcomeres elongated in direct proportion to the amount of stretch. Thus, in both passive and active myofibrils, straightening of Ig domains was accompanied by extension of PEVK which suggests that antibody labeling increased the force required to extend the Ig domains (Fig. 6)

Reproduced with permission from Powers et al. (2016). Copyright 2016, The Company of Biologists Ltd
Activation increases titin-based force and stiffness of wild type psoas myofibrils, but not mdm myofibrils, during stretch. Stress (nN/μm2) versus sarcomere length (μm) of single psoas myofibrils from wild type (WT, black open symbols) and muscular dystrophy with myositis (mdm, gray filled symbols) mice during active (diamonds) and passive stretch (circles). There is no difference in passive stress between WT and mdm myofibrils. Actively stretched WT myofibrils are stiffer than active mdm and both passive mdm and WT, while actively stretched mdm myofibrils do not differ in stiffness from passively stretched mdm and WT myofibrils
.
While the data from most actively stretched myofibrils activated at SL ~ 1.5 μm were similar to the passive data (Fig. 5d), in one myofibril all of the sarcomeres (n = 7) showed the pattern expected if N2A binds to actin (Dutta et al. 2018) and prevents extension of the proximal segment. In this myofibril, which was activated at SL > 2.0 μm, the length of the proximal segment remained constant up to SL > 4.0 μm.
The increased force required to straighten Ig domains in passive stretch data suggests that labeling with the N2A antibody increased the stiffness of the proximal Ig domains (Linke et al. 1996), likely due to secondary antibody cross-linking (Shimamoto et al. 2009; DuVall 2015). This cross-linking effect could also prevent N2A binding to actin, which would also explain the active stretch results that extension of proximal Ig domains and PEVK occurred simultaneously at short sarcomere lengths (< 2.4 μm). DuVall (2015) observed that labeling with secondary, but not primary F146, antibodies increased force during passive stretch, consistent with secondary antibody cross-linking. The observation that most myofibrils were stretched from very short initial lengths (~ 1.5 μm) and that the myofibril exhibiting no proximal extension was stretched from a longer initial sarcomere length > 2.0 μm suggest that cross-linking of secondary antibodies might be reduced or prevented by stretching sarcomeres from a longer initial SL. Based on these preliminary results, our ongoing work is focused on using labeled primary N2A antibodies in single skinned muscle fibers to further investigate titin–actin interactions in muscle sarcomeres. Single skinned fibers are easy to isolate without damage, and we retain the ability to image epitope elongation during passive and active stretch in individual sarcomeres while gaining greater control over length and activation.
Ca2+-dependent N2A–actin interactions: theoretical implications
If N2A titin, situated at the distal end of the proximal tandem Ig domains, binds to thin filaments upon calcium influx in muscle sarcomeres (Fig. 7), then two mechanical consequences are predicted. First, the length of titin’s freely extensible I-band must decrease upon activation (Leonard and Herzog 2010; Nishikawa et al. 2012) because the bound N2A would allow only the PEVK region to extend during active stretch (Fig. 7, right). Second, because titin stiffness in passive muscle is limited by low-force straightening of the proximal tandem Ig domains (Linke et al. 1998b), binding between thin filaments and N2A titin would increase titin stiffness in active muscles, as only the relatively stiffer PEVK region would extend during stretch (Fig. 7, (Nishikawa et al. 2012). Failure of proximal tandem Ig domains to straighten during active stretch would shift the force-extension curve leftward compared to the passive, low-calcium condition (Monroy et al. 2017; Nishikawa 2016). Indeed, there is experimental support for both of these predictions in wild type skeletal muscles and both fail to be observed in skeletal muscles of mdm mice with a 53 amino acid deletion in Ig83 of the N2A region.

Reproduced with permission from Nishikawa (2016). Copyright 2016, The Company of Biologists Ltd
N2A–actin interaction eliminates low-force straightening of tandem Ig domains. a Passive sarcomere at slack length. b As the sarcomere is stretched passively beyond its slack length, the proximal tandem Ig segments unfold approximately to their contour length. c After the proximal tandem Ig segments have reached their contour length, further passive stretching extends the PEVK segment. Adapted from Granzier and Labeit (2004). c Upon activation, N2A titin binds to actin. d When the active sarcomere is stretched, only the PEVK segment (green) extends
In contrast to skeletal muscles, N2A exons are not expressed in the cardiac N2B isoform of titin (Neagoe et al. 2003), which predominates in the hearts of rats and mice. Therefore, the mechanical effects of binding between N2A titin and thin filaments are expected to be absent in cardiac sarcomeres that predominantly express the N2B isoform (Nishikawa et al. 2018a, 2012). Cardiac titin isoforms are much shorter than skeletal muscle isoforms (Neagoe et al. 2003), and their passive length-tension curves are shifted far to the left relative to skeletal muscle isoforms. The ability of skeletal muscles to modulate titin stiffness from the passive to the active state using Ca2+-dependent N2A–actin binding would provide an important benefit for skeletal muscles, which operate in antagonistic pairs. High passive stiffness would limit stretch of muscles by antagonists, adding to contraction loads and reducing efficiency. In contrast, cardiac muscle operates against a pressure load but apparently has no active antagonism and therefore can be tuned to a higher passive stiffness.
A third prediction is that, if N2A titin binds to the nearest available actin subunit upon calcium influx (Nishikawa et al. 2012, 2018a), then titin’s spring properties should also be independent of sarcomere length at the time of activation. Once N2A binds to actin, the slack tandem Ig region can no longer extend during stretch. The only part that will stretch is the PEVK region, and this will be true regardless of where N2A binds to actin. In this way, titin stiffness could increase with activation even at very short sarcomere lengths on the ascending limb of the force–length relationship. This property might help to explain why skeletal but not cardiac muscles have a plateau in the force–length relationship, even though both types of sarcomeres possess a bare zone with no cross bridges on the thick filaments (Allen and Kentish 1985) to which the plateau in force is typically attributed (Gordon et al. 1966). It also might help to explain why the maximum shortening velocity of muscle (Vmax) is nearly constant over a wide range of sarcomere lengths ranging from 1.6 to 2.6 μm (Edman 1978). If cross bridges alone are responsible for muscle shortening, one would expect a proportional relationship between force and Vmax. Indeed, reducing activation either in vivo (Chow and Darling 1999) or in vitro (Gilliver et al. 2011) decreases Vmax. Thus, it is curious that Vmax should be similar at optimal length (2.6 μm) and on the ascending limb of the force–length relationship where force is substantially reduced.
Titin–actin interactions: evidence from mechanical behavior of muscle
It has been argued previously that Ca2+-dependent titin–actin interactions provide a relatively simple explanation for history-dependent muscle properties, including residual force enhancement (Leonard and Herzog 2010; Nishikawa 2016) and force depression (Nishikawa et al. 2012, 2018a, b; Schappacher-Tilp et al. 2015). Indeed, both force enhancement and force depression are negligible in skeletal muscles from mdm mice compared to passive tension at the final length (Tahir et al. 2019). Here, we instead focus on observational evidence that titin’s equilibrium length decreases and titin stiffness increases upon activation.
Muscle equilibrium length decreases upon activation, but not in mdm
Several observations on elastic recoil of active muscle during rapid unloading are consistent with the prediction from Ca2+-dependent N2A–actin binding that the equilibrium length of I-band titin should decrease upon activation. Prior to the discovery of titin, it was understandable that all of the instantaneous elasticity of muscle was thought to reside in the cross bridges (Huxley and Simmons 1971b), and hence that that cross bridges alone account for all of the increased force during active stretch (Lombardi and Piazzesi 1990; Piazzesi and Lombardi 1995). However, a more recent study (Linari et al. 2003) estimated the contribution of cross bridges to energy storage during active muscle stretch at only 12%, with cross bridge elasticity accounting for a mere 2% of the stored energy.
When frog muscles activated at optimal length are rapidly unloaded, they recoil elastically by up to 20% of their length, ~ 77 nm per half sarcomere (Lappin et al. 2006). Similarly, although single fibers from the telson muscle of horseshoe crabs recoil by only 6% of their length during rapid unloading, they shorten by 210 nm per half-sarcomere due to the extremely long length (7 μm) of the sarcomeres in these muscles (Sugi et al. 2000). The large stretch of elastic elements in these muscles far exceeds any plausible limit in the cross bridges or filament lattice (Lappin et al. 2006).
Monroy et al. (2017) found that equilibrium length after elastic recoil was 15% shorter in active than in passive mouse soleus muscles, and there was a 2.9-fold increase in the slope of the stress–strain relationship during unloading (Fig. 8). In contrast, soleus muscles from mdm mice showed no change in equilibrium length and no effect of activation on the slope or intercept of the stress–strain relationship during unloading. These results are consistent with the idea that Ca2+-dependent binding of titin to actin reduces titin free length and increases titin stiffness, but not in mdm muscles with a deletion of ~ 53 amino acid residues in Ig83 within the N2A region.

Reproduced with permission from Monroy et al. (2017). Copyright 2017, The Company of Biologists Ltd
Activation decreases equilibrium length and increases stiffness of wild type soleus muscles, but not mdm soleus, during rapid unloading. Stress–strain curves for activated (solid symbols) and passively stretched (open symbols) wild type (a) and mdm soleus (b) during rapid unloading from the same initial stress
Due to potentially confounding effects of the extracellular matrix and other elastic structures in intact muscles, Monroy et al. (2017) repeated their experiments at 85% L0 to fully unload series elastic elements outside the sarcomeres and thereby reduce their contribution to elastic recoil. The results demonstrated no measurable contribution of extrinsic elastic elements to elastic recoil during rapid unloading at L0, consistent with recent experiments demonstrating that titin contributes more to passive stiffness than ECM even in intact muscles (Brynnel et al. 2018).
Titin force and stiffness increase upon activation, but not in mdm
Recent studies provide evidence that titin force and stiffness increase in calcium-activated myofibrils. Leonard and Herzog (2010) demonstrated that titin force and stiffness are greater in actively stretched than in passively stretched single rabbit psoas myofibrils, when myofibrils were stretched beyond overlap of the thick and thin filaments where no contribution of cross bridges to force or stiffness is possible. Subsequent studies (Powers et al. 2014) confirmed the basic result in mouse psoas. Although Ca2+ alone also increases titin stiffness (Labeit et al. 2003), the effect is too small to account for the increase in titin stiffness that occurs upon activation (Leonard and Herzog 2010). Significantly, the mdm mutation prevents the increase in titin-based stiffness (Powers et al. 2016) that normally occurs upon activation of wild type myofibrils (Leonard and Herzog 2010; Powers et al. 2014).
Other laboratories have found increases in titin stiffness (Rassier 2017) which are smaller in magnitude, likely attributable to use of different methods (Herzog 2017). Although the results have also been criticized on the grounds that the large titin-based forces observed in Leonard and Herzog’s (2010) study may be due to detachment of A-band titin from myosin at long sarcomere lengths (Freundt and Linke 2018), if such a detachment occurred, it would reduce rather than increase titin-based force in the myofibrils.
Muscle force depends on sarcomere length and velocity at activation, but not in mdm
Muscles can function as motors, brakes, springs or struts (Dickinson et al. 2000; Ahn et al. 2003; George et al. 2012). In work loop experiments (Josephson 1985), a muscle can be activated at different phases of a stretch–shortening cycle. The mechanical properties of a muscle are determined by the phase within the cycle at which the muscle is activated (Nishikawa et al. 2018b). When stretched passively to long lengths and activated during shortening, muscles function as motors that do positive work (Ahn et al. 2003). When activated at short lengths at the onset of active stretch, muscles act as brakes that absorb work (Hessel and Nishikawa 2017). When activated at intermediate lengths and phases, muscles function as springs or struts and perform relatively little net work per cycle (Ahn et al. 2003).
In an example work loop experiment (Fig. 9), when extensor digitorum longus (EDL) muscles from wild type mice are stimulated during lengthening, force rises rapidly as work is absorbed (Fig. 9a). When stimulated during shortening after passive stretch, wild type EDL muscles perform positive work (Fig. 9b). In contrast, over the same relative stretch–shortening cycle with the same phase of stimulation, mdm EDL muscles absorb work regardless of their length at the onset of activation (Fig. 9c, d). These results are consistent with the decrease in equilibrium length and increase in active stiffness predicted by Ca2+-dependent N2A–actin binding, and further demonstrate the importance of N2A titin in determining the mechanical function of active muscle.

The mechanical properties of muscles are determined by the length and velocity within a stretch–shortening cycle at which they are activated. a When a wild type extensor digitorum longus (EDL) muscle is stimulated during lengthening, the force rises rapidly and work is absorbed (clock-wise cycle). b When stretched passively and activated during shortening, the EDL functions as a motor that performs work (counter-clockwise cycle). In contrast, the mdm soleus absorbs work regardless of its length at the onset of activation (c, d). Muscles were subjected to ± 5% cyclical length changes at 5 Hz while stimulated submaximally at 50 Hz (four stimuli per cycle). White plus symbols indicate the first stimulus and filled circles indicate subsequent stimuli. Arrows indicate the direction of the work loop
Accepted functions of titin in active muscle require high stiffness
Several functions for titin in active muscle are now relatively well accepted, including the idea that titin maintains sarcomere integrity in both longitudinal and radial directions. Horowits et al. (1986) and Horowits and Podolsky (1987) suggested that titin maintains the longitudinal alignment of thick filaments in the center of muscle sarcomeres (Horowits et al. 1986; Horowits and Podolsky 1987). They showed that, in sarcomeres exposed to ionizing radiation to degrade titin, the alignment of thick filaments at the center of the sarcomere was lost as individual thick filaments moved toward one or the other of the z-discs upon activation. Although the experiments were initially criticized on the grounds that ionizing radiation may have affected other proteins in addition to titin, the idea that titin is responsible for this phenomenon is now increasingly accepted (Linke 2018).
Due to the geometry of muscle sarcomeres, in which titin molecules extend from the end of the thick filaments to the thin filaments near the Z-disc, longitudinal forces in titin that maintain alignment of the thick filaments must also exert forces in the radial direction within a sarcomere. Indeed, a recent study suggests that myofibrils from rabbit psoas muscles with a stiffer titin isoform have higher radial forces in the A-band after stretch than rabbit diaphragm with a more compliant titin isoform (Li et al. 2016). While Li et al. (2016) proposed that the increasing stiffness of the A-band could result from sensing of titin stiffness by other proteins in the A-band, the increased radial forces they observed must necessarily be related mechanically to the longitudinal force and stiffness of titin.
Evidence from X-ray diffraction in skeletal muscles (Irving et al. 2011; Ma et al. 2018a) further suggests that titin contributes, directly or indirectly, to longitudinal strain of thick and thin filaments that occurs during passive and active stretch of muscle fibers. Because six titin filaments are associated with each thick filament in the A band and attach to the thin filaments in the Z-disc (Trinick 1996), passive stretch of titin would also stretch the thick filaments. Indeed, thick filaments can be seen to taper at the edge of the A-band during passive stretch to very long sarcomere lengths (Locker and Leet 1975). The observation that the non-linear force-extension curve of thick filaments is similar during passive and active stretch (Ma et al. 2018b) suggests a role for titin, whether direct or indirect, under both conditions.
The observations that: (i) ionizing radiation degrades titin and results in loss of thick filament alignment within the A-band (Horowits et al. 1986); (ii) titin isoforms which differ in length and stiffness have differing radial stiffness in the A-band (Li et al. 2016); (iii) titin-based passive tension is associated with thick filament strain during passive stretch (Irving et al. 2011); and (iv) force extension curves of thick filaments during passive and active stretch are similar (Ma et al. 2018b) all suggest a role for titin in transmitting and distributing forces within muscle sarcomeres under a wide variety of conditions, although it is debated whether this role is direct or indirect (Li et al. 2016; Linke 2018).
Because titin stiffness is limited by straightening of proximal tandem Ig domains in the I-band under low-force (Linke et al. 1998a, b), a mechanism such as Ca2+-dependent binding of N2A titin to actin is a theoretical requirement for titin to play even an indirect role in balancing destabilizing forces within sarcomeres during active stretch. Without such a mechanism, the stabilizing functions of titin should only be apparent at longer sarcomere lengths above ~ 3.0 μm. At shorter lengths, destabilizing forces would merely deform the proximal tandem Ig region and neither longitudinal stabilization of thick filaments nor radial stabilization of lattice spacing would be possible. Even mechanisms such as titin stiffness sensing by A-band proteins (Li et al. 2016) would be impossible at shorter sarcomere lengths without stabilization of titin’s compliant proximal tandem Ig domains.
A cautionary tale: twitchin–actin interactions in catch muscle
The “catch” phenomenon of invertebrate muscles shares several similarities with residual force enhancement in vertebrate skeletal muscle (Sugi and Suzuki 1978), including: (1) resistance to stretch (for invertebrate catch, (Butler and Siegman 2010); for skeletal muscle, (Abbott and Aubert 1952); (2) maintenance of force at low energetic cost (for invertebrate catch, (Baguet and Gillis 1968); for skeletal muscle, (Bigland-Ritchie and Woods 1976; Ortega et al. 2015); and (3) that persists for an extended time (for invertebrate catch, (Butler and Siegman 2010); for skeletal muscle, (Herzog et al. 2003). Because they are likely to have evolved independently (Lindstedt and Nishikawa 2017), there are numerous differences as well. For example, there is an absence of force redevelopment after unloading in catch (Jewell 1959) but not in force enhancement (Lee et al. 2001).
Although once attributed to cross-bridges (Tameyasu and Sugi 1976), myosin light-chain phosphorylation (Takahashi et al. 1988), and a variety of other mechanisms [see (Hooper et al. 2008) and references therein], catch is now thought to result from binding between actin and dephosphorylated twitchin (Yamada et al. 2001), a giant sarcomeric protein orthologous to titin (Kenny et al. 1999).
The history of the mechanism of invertebrate catch (Hooper et al. 2008) provides insights into the role of giant proteins in muscle contraction and suggests experimental approaches that may help to resolve other mechanisms. In molluscan catch, an elastic element develops upon muscle activation (Butler et al. 2010). This elastic element persists for long periods after deactivation (Butler et al. 2010). Until 1998, when Butler and Siegman first observed that the phosphorylation state of twitchin was correlated with the catch state (Butler et al. 1998), nobody had even suggested that twitchin might play a role.
A potential pitfall of the bottom-up approach—in which biochemical pathways are identified in vitro or in vivo and are then invoked to explain higher-level phenomena—is that a multitude of pathways may exist. Yet, only one or a few pathways are likely to contribute substantially to any given phenomenon. In the catch state of invertebrate muscles, no fewer than 26 proteins change their phosphorylation state (Hooper et al. 2008). In the case of catch, the twitchin–actin binding mechanism was only accepted as the mechanism of catch after Yamada et al. (2001) demonstrated that catch could be observed in an in vitro assay containing only myosin, actin and twitchin, and that catch depended only on the phosphorylation state of twitchin. Later studies demonstrated that Ca2+ influx triggers dephosphorylation of twitchin (Funabara et al. 2005), and that binding of dephosphorylated twitchin to actin is sufficient to explain catch (Shelud’ko et al. 2004). Thus, the mere existence of biochemical pathways in vivo and even the correlation with mechanical events, is insufficient to demonstrate causation.
Deductive reasoning: other examples from muscle biophysics
On the other hand, experimental observations frequently appear to require the existence of a new mechanism long before it can be demonstrated to exist experimentally—the top-down approach. Often, this theoretical necessity provides the motivation to search for mechanisms. However, many examples also exist in which the logic of a theory or the strength of the evidence was questioned, and only later did provision of a mechanism justify acceptance of the original deduction. The history of the concept of continental drift, fraught with a firestorm of vitriolic criticism and ridicule from skeptics of the time whose research assumed an unchanging Earth, is a well-known example (Romano and Cifelli 2015). In retrospect, after the mechanism of sea floor spreading was discovered, Wegener’s extensive geological, geophysical and paleontological evidence seems remarkably persuasive.
Two examples from the history of muscle research further illustrate the power of deductive reasoning to generate mechanistic hypotheses. The sliding filament hypothesis itself met with considerable initial skepticism from leading scientists of the time, including Albert Szent-Györgyi and Paul Flory (Maruyama 1995). Despite the observational evidence for sliding filaments—that the A-band length remained constant during muscle contraction whereas the I-band and H-band decreased in length (Hanson and Huxley 1953)—Maruyama (1995) vividly recalls Jean Hanson at a symposium on biomacromolecules in the early 1960’s shouting, “I know I cannot explain the mechanism yet, but the sliding is a fact”. Of course, it is now well accepted that ATP-hydrolyzing acto-myosin cross bridges (Huxley 1957) provide the chemical energy for sliding of thin filaments toward the M-line during active muscle contraction.
The second example from muscle research involves the so-called “S-filaments” of Hanson & Huxley (Dos Remedios and Gilmour 2017; Hanson and Huxley 1953; Hitchcock-DeGregori and Irving 2014; Maruyama 1995), whose existence appeared to be required by the experimental observation that the structure and elasticity of myofibrils is maintained even after myosin and actin have been extracted (Huxley and Hanson 1954). Yet, when initial evidence from electron microscopy appeared to indicate the existence of these filaments (Hoyle 1967; Sjostrand 1962), Huxley (1964) and Hanson (1968) were among the most ardent skeptics. Eventually, Maruyama (1976) and others (Wang et al. 1979) were able to purify the titin/connectin protein from muscles and to demonstrate its existence and spatial localization within sarcomeres (Furst et al. 1988). A similar history of skepticism was until recently associated with the idea that titin plays a role in active muscle (Linke 2018).
Recognizing that it is essential to test whether Ca2+-dependent binding between N2A titin and actin, as observed in vitro using protein constructs, also occurs in vivo within muscle sarcomeres, efforts are currently underway using labeling with N2A and other anti-titin antibodies to test this hypothesis. In the meantime, extensive evidence from numerous experiments demonstrates that skeletal muscles behave in a manner that can be explained by Ca2+-dependent binding of N2A titin to actin. Given the new in vitro evidence for Ca2+-dependent binding between N2A titin and actin, it is time to acknowledge the evidence for such interactions from the known mechanical behavior of muscle.
References
Abbott BC, Aubert XM (1952) The force exerted by active striated muscle during and after change of length. J Physiol 117:77–86
Ahn AN, Monti RJ, Biewener AA (2003) In vivo and in vitro heterogeneity of segment length changes in the semimembranosus muscle of the toad. J Physiol 549:877–888. https://doi.org/10.1113/jphysiol.2002.038018
Allen DG, Kentish JC (1985) The cellular basis of the length-tension relation in cardiac muscle. J Mol Cell Cardiol 17:821–840
Baguet F, Gillis JM (1968) Energy cost of tonic contraction in a lamellibranch catch muscle. J Physiol 198:127–143. https://doi.org/10.1113/jphysiol.1968.sp008597
Bianco P, Nagy A, Kengyel A, Szatmari D, Martonfalvi Z, Huber T, Kellermayer MS (2007) Interaction forces between F-actin and titin PEVK domain measured with optical tweezers. Biophys J 93:2102–2109. https://doi.org/10.1529/biophysj.107.106153
Bigland-Ritchie B, Woods JJ (1976) Integrated electromyogram and oxygen uptake during positive and negative work. J Physiol 260:267–277. https://doi.org/10.1113/jphysiol.1976.sp011515
Brynnel A et al (2018) Downsizing the molecular spring of the giant protein titin reveals that skeletal muscle titin determines passive stiffness and drives longitudinal hypertrophy. Elife 7:150. https://doi.org/10.7554/eLife.40532
Butler TM, Siegman MJ (2010) Mechanism of catch force: tethering of thick and thin filaments by twitchin. J Biomed Biotechnol 2010:725207. https://doi.org/10.1155/2010/725207
Butler TM, Mooers SU, Li C, Narayan S, Siegman MJ (1998) Regulation of catch muscle by twitchin phosphorylation: effects on force, ATPase, and shortening. Biophys J 75:1904–1914. https://doi.org/10.1016/S0006-3495(98)77631-3
Butler TM, Mooers SU, Narayan SR, Siegman MJ (2010) The N-terminal region of twitchin binds thick and thin contractile filaments: redundant mechanisms of catch force maintenance. J Biol Chem 285:40654–40665. https://doi.org/10.1074/jbc.M110.166041
Chow JW, Darling WG (1999) The maximum shortening velocity of muscle should be scaled with activation. J Appl Physiol 86:1025–1031. https://doi.org/10.1152/jappl.1999.86.3.1025
Cornachione AS, Leite F, Bagni MA, Rassier DE (2016) The increase in non-cross-bridge forces after stretch of activated striated muscle is related to titin isoforms. Am J Physiol Cell Physiol 310:C19–C26. https://doi.org/10.1152/ajpcell.00156.2015
Dick TJM, Biewener AA, Wakeling JM (2017) Comparison of human gastrocnemius forces predicted by Hill-type muscle models and estimated from ultrasound images. J Exp Biol 220:1643–1653. https://doi.org/10.1242/jeb.154807
Dickinson MH, Farley CT, Full RJ, Koehl MAR, Kram R, Lehman S (2000) How animals move: an integrative view. Science 288:100–106
Dos Remedios C, Gilmour D (2017) An historical perspective of the discovery of titin filaments. Biophys Rev 9:179–188. https://doi.org/10.1007/s12551-017-0269-3
Dutta S et al (2018) Calcium increases titin N2A binding to F-actin and regulated thin filaments. Sci Rep 8:14575. https://doi.org/10.1038/s41598-018-32952-8
DuVall M (2015) Titin regulation of active and passive force in skeletal muscle. Ph.D. Dissertation, University of Calgary, Canada
DuVall MM, Jinha A, Schappacher-Tilp G, Leonard TR, Herzog W (2017) Differences in titin segmental elongation between passive and active stretch in skeletal muscle. J Exp Biol 220:4418–4425. https://doi.org/10.1242/jeb.160762
Edman KA (1978) Maximum velocity of shortening in relation to sarcomere length and degree of activation of frog muscle fibres. J Physiol 278:9P–10P
Fenn WO (1923) A quantitative comparison between the energy liberated and the work performed by the isolated sartorius muscle of the frog. J Physiol 58:175–203. https://doi.org/10.1113/jphysiol.1923.sp002115
Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S (2000) Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res 86:1114–1121
Freundt JK, Linke WA (2018) Titin as a force generating muscle protein under regulatory control. J Appl Physiol 126(5):1474–1482. https://doi.org/10.1152/japplphysiol.00865.2018
Funabara D, Kanoh S, Siegman MJ, Butler TM, Hartshorne DJ, Watabe S (2005) Twitchin as a regulator of catch contraction in molluscan smooth muscle. J Muscle Res Cell Motil 26:455–460. https://doi.org/10.1007/s10974-005-9029-2
Furst DO, Osborn M, Nave R, Weber K (1988) The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: a map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol 106:1563–1572
Garvey SM, Rajan C, Lerner AP, Frankel WN, Cox GA (2002) The muscular dystrophy with myositis (mdm) mouse mutation disrupts a skeletal muscle-specific domain of titin. Genomics 79:146–149. https://doi.org/10.1006/geno.2002.6685
Gautel M, Djinovic-Carugo K (2016) The sarcomeric cytoskeleton: from molecules to motion. J Exp Biol 219:135–145. https://doi.org/10.1242/jeb.124941
George NT, Sponberg S, Daniel TL (2012) Temperature gradients drive mechanical energy gradients in the flight muscle of Manduca sexta. J Exp Biol 215:471–479. https://doi.org/10.1242/jeb.062901
Gilliver SF, Jones DA, Rittweger J, Degens H (2011) Variation in the determinants of power of chemically skinned type I rat soleus muscle fibres. J Comp Physiol A 197:311–319. https://doi.org/10.1007/s00359-010-0613-6
Gordon AM, Huxley AF, Julian FJ (1966) The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 184:170–192
Granzier HL, Labeit S (2004) The giant protein titin: a major player in myocardial mechanics, signaling, and disease. Circ Res 94:284–295. https://doi.org/10.1161/01.RES.0000117769.88862.F8
Gregorio CC et al (1998) The NH2 terminus of titin spans the Z-disc: its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol 143:1013–1027
Gregorio CC, Granzier H, Sorimachi H, Labeit S (1999) Muscle assembly: a titanic achievement. Curr Opin Cell Biol 11:18–25
Hanson J (1968) Recent x-ray diffraction studies of muscle. Q Rev Biophys 1:177–216
Hanson J, Huxley HE (1953) Structural basis of the cross-striations in muscle. Nature 172:530–532
Herzog W (2017) Skeletal muscle mechanics: questions, problems and possible solutions. J Neuroeng Rehabil 14:98. https://doi.org/10.1186/s12984-017-0310-6
Herzog W, Schachar R, Leonard TR (2003) Characterization of the passive component of force enhancement following active stretching of skeletal muscle. J Exp Biol 206:3635–3643
Hessel AL, Nishikawa KC (2017) Effects of a titin mutation on negative work during stretch-shortening cycles in skeletal muscles. J Exp Biol 220:4177–4185. https://doi.org/10.1242/jeb.163204
Hill AV (1922) The maximum work and mechanical efficiency of human muscles, and their most economical speed. J Physiol 56:19–41. https://doi.org/10.1113/jphysiol.1922.sp001989
Hitchcock-DeGregori SE, Irving TC, Hugh E (2014) Huxley: the compleat biophysicist. Biophys J 107:1493–1501. https://doi.org/10.1016/j.bpj.2014.07.069
Hooper SL, Hobbs KH, Thuma JB (2008) Invertebrate muscles: thin and thick filament structure; molecular basis of contraction and its regulation, catch and asynchronous muscle. Prog Neurobiol 86:72–127. https://doi.org/10.1016/j.pneurobio.2008.06.004
Horowits R, Podolsky RJ (1987) The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: evidence for the role of titin filaments. J Cell Biol 105:2217–2223
Horowits R, Kempner ES, Bisher ME, Podolsky RJ (1986) A physiological role for titin and nebulin in skeletal muscle. Nature 323:160–164. https://doi.org/10.1038/323160a0
Houmeida A, Holt J, Tskhovrebova L, Trinick J (1995) Studies of the interaction between titin and myosin. J Cell Biol 131:1471–1481
Hoyle G (1967) Diversity of striated muscle. Am Zool 7:435–449
Huxley AF (1957) Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7:255–318
Huxley HE (1964) Structural arrangements and the contraction mechanism in striated muscle. Proc R Soc Lond B 160:442–448. https://doi.org/10.1098/rspb.1964.0054
Huxley AF (1973) A note suggesting that the cross-bridge attachment during muscle contraction may take place in two stages. Proc R Soc Lond B 183:83–86. https://doi.org/10.1098/rspb.1973.0006
Huxley H, Hanson J (1954) Changes in the cross-striations of muscle during contraction and stretch and their structural interpretation. Nature 173:973–976
Huxley AF, Simmons RM (1971a) Mechanical properties of the cross-bridges of frog striated muscle. J Physiol 218(Suppl):59P–60P
Huxley AF, Simmons RM (1971b) Proposed mechanism of force generation in striated muscle. Nature 233:533–538
Irving M (2017) Regulation of contraction by the thick filaments in skeletal muscle. Biophys J 113:2579–2594. https://doi.org/10.1016/j.bpj.2017.09.037
Irving T, Wu Y, Bekyarova T, Farman GP, Fukuda N, Granzier H (2011) Thick-filament strain and interfilament spacing in passive muscle: effect of titin-based passive tension. Biophys J 100:1499–1508. https://doi.org/10.1016/j.bpj.2011.01.059
Jewell BR (1959) The nature of the phasic and the tonic responses of the anterior byssal retractor muscle of Mytilus. J Physiol 149:154–177. https://doi.org/10.1113/jphysiol.1959.sp006332
Jin JP (1995) Cloned rat cardiac titin class I and class II motifs: expression, purification, characterization, and interaction with F-actin. J Biol Chem 270:6908–6916
Jin JP (2000) Titin-thin filament interaction and potential role in muscle function. Adv Exp Med Biol 481:319–333 discussion 334–315
Josephson RK (1985) Mechanical power output from striated muscle during cyclic contraction. J Exp Biol 114:493–512
Kellermayer MS, Granzier HL (1996) Calcium-dependent inhibition of in vitro thin-filament motility by native titin. FEBS Lett 380:281–286
Kenny PA, Liston EM, Higgins DG (1999) Molecular evolution of immunoglobulin and fibronectin domains in titin and related muscle proteins. Gene 232:11–23
Konhilas JP, Irving TC, de Tombe PP (2002) Myofilament calcium sensitivity in skinned rat cardiac trabeculae: role of interfilament spacing. Circ Res 90:59–65
Kulke M et al (2001) Interaction between PEVK-titin and actin filaments: origin of a viscous force component in cardiac myofibrils. Circ Res 89:874–881
Labeit S, Kolmerer B (1995) Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 270:293–296
Labeit S, Gautel M, Lakey A, Trinick J (1992) Towards a molecular understanding of titin. EMBO J 11:1711–1716
Labeit D et al (2003) Calcium-dependent molecular spring elements in the giant protein titin. Proc Natl Acad Sci USA 100:13716–13721. https://doi.org/10.1073/pnas.2235652100
Lappin AK, Monroy JA, Pilarski JQ, Zepnewski ED, Pierotti DJ, Nishikawa KC (2006) Storage and recovery of elastic potential energy powers ballistic prey capture in toads. J Exp Biol 209:2535–2553. https://doi.org/10.1242/jeb.02276
Lee HD, Herzog W, Leonard T (2001) Effects of cyclic changes in muscle length on force production in in situ cat soleus. J Biomech 34:979–987
Lee SS, Arnold AS, Miara Mde B, Biewener AA, Wakeling JM (2013) Accuracy of gastrocnemius muscles forces in walking and running goats predicted by one-element and two-element Hill-type models. J Biomech 46:2288–2295. https://doi.org/10.1016/j.jbiomech.2013.06.001
Leonard TR, Herzog W (2010) Regulation of muscle force in the absence of actin-myosin-based cross-bridge interaction. Am J Physiol Cell Physiol 299:C14–C20. https://doi.org/10.1152/ajpcell.00049.2010
Li Q, Jin JP, Granzier HL (1995) The effect of genetically expressed cardiac titin fragments on in vitro actin motility. Biophys J. 69:1508–1518. https://doi.org/10.1016/S0006-3495(95)80021-4
Li Y, Lang P, Linke WA (2016) Titin stiffness modifies the force-generating region of muscle sarcomeres. Sci Rep 6:24492. https://doi.org/10.1038/srep24492
Li Y, Unger A, von Frieling-Salewsky M, Rivas Pardo JA, Fernandez JM, Linke WA (2018) Quantifying the titin contribution to muscle force generation using a novel method to specifically cleave the titin springs in situ. Biophys J 114:645a. https://doi.org/10.1016/j.bpj.2017.11.3480
Linari M, Woledge RC, Curtin NA (2003) Energy storage during stretch of active single fibres from frog skeletal muscle. J Physiol 548:461–474. https://doi.org/10.1113/jphysiol.2002.032185
Lindstedt S, Nishikawa K (2017) Huxleys’ missing filament: form and function of titin in vertebrate striated muscle. Ann Rev Physiol 79:145–166. https://doi.org/10.1146/annurev-physiol-022516-034152
Linke WA, Ivemeyer M, Olivieri N, Kolmerer B, Ruegg JC, Labeit S (1996) Towards a molecular understanding of the elasticity of titin. J Mol Biol 261:62–71
Linke WA (2018) Titin gene and protein functions in passive and active muscle. Annu Rev Physiol 80:389–411. https://doi.org/10.1146/annurev-physiol-021317-121234
Linke WA, Ivemeyer M, Labeit S, Hinssen H, Ruegg JC, Gautel M (1997) Actin-titin interaction in cardiac myofibrils: probing a physiological role. Biophys J 73:905–919. https://doi.org/10.1016/S0006-3495(97)78123-2
Linke WA, Ivemeyer M, Mundel P, Stockmeier MR, Kolmerer B (1998a) Nature of PEVK-titin elasticity in skeletal muscle. Proc Natl Acad Sci USA 95:8052–8057
Linke WA, Stockmeier MR, Ivemeyer M, Hosser H, Mundel P (1998b) Characterizing titin’s I-band Ig domain region as an entropic spring. J Cell Sci 111:1567–1574
Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC (1999) I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 146:631–644
Linke WA et al (2002) PEVK domain of titin: an entropic spring with actin-binding properties. J Struct Biol 137:194–205. https://doi.org/10.1006/jsbi.2002.4468
Locker RH, Leet NG (1975) Histology of highly-stretched beef muscle. I. The fine structure of grossly stretched single fibers. J Ultrastruct Res 52:64–75
Lombardi V, Piazzesi G (1990) The contractile response during steady lengthening of stimulated frog muscle fibres. J Physiol 431:141–171
Lombardi V, Piazzesi G, Ferenczi MA, Thirlwell H, Dobbie I, Irving M (1995) Elastic distortion of myosin heads and repriming of the working stroke in muscle. Nature 374:553–555. https://doi.org/10.1038/374553a0
Ma W, Gong H, Irving T (2018a) Myosin head configurations in resting and contracting murine skeletal muscle. Int J Mol Sci 19:2643. https://doi.org/10.3390/ijms19092643
Ma W, Gong H, Kiss B, Lee EJ, Granzier H, Irving T (2018b) Thick-filament extensibility in intact skeletal muscle. Biophys J 115:1580–1588. https://doi.org/10.1016/j.bpj.2018.08.038
Maruyama K (1976) Connectin, an elastic protein from myofibrils. J Biochem 80:405–407
Maruyama K (1995) Birth of the sliding filament concept in muscle contraction. J Biochem 117:1–6
Maruyama K, Natori R, Nonomura Y (1976) New elastic protein from muscle. Nature 262:58–60
Maruyama K, Hu DH, Suzuki T, Kimura S (1987) Binding of actin filaments to connectin. J Biochem 101:1339–1346
Mijailovich SM, Stojanovic B, Nedic D, Svicevic M, Geeves MA, Irving TC, Granzier HL (2019) Nebulin and titin modulate cross-bridge cycling and length-dependent calcium sensitivity. J Gen Physiol 151:680–704. https://doi.org/10.1085/jgp.201812165
Monroy JA, Powers KL, Pace CM, Uyeno T, Nishikawa KC (2017) Effects of activation on the elastic properties of intact soleus muscles with a deletion in titin. J Exp Biol 220:828–836. https://doi.org/10.1242/jeb.139717
Murayama T, Nakauchi Y, Kimura S, Maruyama K (1989) Binding of connectin to myosin filaments. J Biochem 105:323–326
Nagy A, Cacciafesta P, Grama L, Kengyel A, Malnasi-Csizmadia A, Kellermayer MS (2004) Differential actin binding along the PEVK domain of skeletal muscle titin. J Cell Sci 117:5781–5789. https://doi.org/10.1242/jcs.01501
Neagoe C, Opitz CA, Makarenko I, Linke WA (2003) Gigantic variety: expression patterns of titin isoforms in striated muscles and consequences for myofibrillar passive stiffness. J Muscle Res Cell Motil 24:175–189
Nishikawa K (2016) Eccentric contraction: unraveling mechanisms of force enhancement and energy conservation. J Exp Biol 219:189–196. https://doi.org/10.1242/jeb.124057
Nishikawa KC, Monroy JA, Uyeno TE, Yeo SH, Pai DK, Lindstedt SL (2012) Is titin a ‘winding filament’? A new twist on muscle contraction. Proc Biol Sci 279:981–990. https://doi.org/10.1098/rspb.2011.1304
Nishikawa K, Lindstedt SL, LaStayo PC (2018a) Basic science and clinical use of eccentric contractions: history and uncertainties. J Sport Health Sci 7:265–274
Nishikawa K, Tahir U, Monroy JA (2018b) Muscle function from organisms to molecules. Int Comp Biol 58:194–206
Obermann WM, Gautel M, Steiner F, van der Ven PF, Weber K, Furst DO (1996) The structure of the sarcomeric M band: localization of defined domains of myomesin, M-protein, and the 250-kD carboxy-terminal region of titin by immunoelectron microscopy. J Cell Biol 134:1441–1453
Ortega JO, Lindstedt SL, Nelson FE, Jubrias SA, Kushmerick MJ, Conley KE (2015) Muscle force, work and cost: a novel technique to revisit the Fenn effect. J Exp Biol 218:2075–2082. https://doi.org/10.1242/jeb.114512
Pfuhl M, Gautel M (2012) Structure, interactions and function of the N-terminus of cardiac myosin binding protein C (MyBP-C): who does what, with what, and to whom? J Muscle Res Cell Motil 33:83–94. https://doi.org/10.1007/s10974-012-9291-z
Piazzesi G, Lombardi V (1995) A cross-bridge model that is able to explain mechanical and energetic properties of shortening muscle. Biophys J 68:1966–1979. https://doi.org/10.1016/S0006-3495(95)80374-7
Powers K, Schappacher-Tilp G, Jinha A, Leonard T, Nishikawa K, Herzog W (2014) Titin force is enhanced in actively stretched skeletal muscle. J Exp Biol 217:3629–3636. https://doi.org/10.1242/jeb.105361
Powers K, Nishikawa K, Joumaa V, Herzog W (2016) Decreased force enhancement in skeletal muscle sarcomeres with a deletion in titin. J Exp Biol 219:1311–1316. https://doi.org/10.1242/jeb.132027
Rassier DE (2017) Sarcomere mechanics in striated muscles: from molecules to sarcomeres to cells. Am J Physiol Cell Physiol 313:C134–C145. https://doi.org/10.1152/ajpcell.00050.2017
Rivas-Pardo JA, Eckels EC, Popa I, Kosuri P, Linke WA, Fernandez JM (2016) Work done by titin protein folding assists muscle contraction. Cell Rep 14:1339–1347. https://doi.org/10.1016/j.celrep.2016.01.025
Romano M, Cifelli RL (2015) Plate tectonics: continental-drift opus turns 100. Nature 526:43. https://doi.org/10.1038/526043e
Sanger JW, Sanger JM (2001) Fishing out proteins that bind to titin. J Cell Biol 154:21–24
Schappacher-Tilp G, Leonard T, Desch G, Herzog W (2015) A novel three-filament model of force generation in eccentric contraction of skeletal muscles. PLoS ONE 10:e0117634. https://doi.org/10.1371/journal.pone.0117634
Shelud’ko NS, Matusovskaya GG, Permyakova TV, Matusovsky OS (2004) Twitchin, a thick-filament protein from molluscan catch muscle, interacts with F-actin in a phosphorylation-dependent way. Arch Biochem Biophys 432:269–277. https://doi.org/10.1016/j.abb.2004.10.006
Shimamoto Y, Suzuki M, Mikhailenko SV, Yasuda K, Ishiwata S (2009) Inter-sarcomere coordination in muscle revealed through individual sarcomere response to quick stretch. Proc Nat Acad Sci USA 106:11954–11959
Sjostrand FS (1962) The connections between A- and I-band filaments in striated frog muscle. J Ultrastruct Res 7:225–246
Soteriou A, Clarke A, Martin S, Trinick J (1993) Titin folding energy and elasticity. Proc Biol Sci 254:83–86. https://doi.org/10.1098/rspb.1993.0130
Sugi H, Suzuki S (1978) Ultrastructural and physiological studies on the longitudinal body wall muscle of Dolabella auricularia. I. Mechanical response and ultrastructure. J Cell Biol 79:454–466
Sugi H, Akimoto T, Kobayashi T, Suzuki S, Shimada M (2000) Possible contribution of titin filaments to the compliant series elastic component in horseshoe crab skeletal muscle fibers. Adv Exp Med Biol 481:371–380 discussion 381–372
Tahir U, Monroy JA, Rice NA, Nishikawa K (2019) Effects of a titin mutation on force enhancement and force depression in mouse soleus muscles. J Exp Biol
Takahashi M, Sohma H, Morita F (1988) The steady state intermediate of scallop smooth muscle myosin ATPase and effect of light chain phosphorylation. A molecular mechanism for catch contraction. J Biochem 104:102–107
Tameyasu T, Sugi H (1976) The series elastic component and the force-velocity relation in the anterior byssal retractor muscle of Mytilus edulis during active and catch contractions. J Exp Biol 64:497–510
Tatsumi R, Maeda K, Hattori A, Takahashi K (2001) Calcium binding to an elastic portion of connectin/titin filaments. J Muscle Res Cell Motil 22:149–162
Trinick J (1996) Interaction of titin/connectin with the thick filament. Adv Biophys 33:81–90
Trombitas K, Granzier H (1997) Actin removal from cardiac myocytes shows that near Z line titin attaches to actin while under tension. Am J Physiol 273:C662–C670
von Castelmur E et al (2008) A regular pattern of Ig super-motifs defines segmental flexibility as the elastic mechanism of the titin chain. Proc Natl Acad Sci USA 105:1186–1191. https://doi.org/10.1073/pnas.0707163105
Wakabayashi K, Sugimoto Y, Tanaka H, Ueno Y, Takezawa Y, Amemiya Y (1994) X-ray diffraction evidence for the extensibility of actin and myosin filaments during muscle contraction. Biophys J 67:2422–2435. https://doi.org/10.1016/S0006-3495(94)80729-5
Wang K, McClure J, Tu A (1979) Titin: major myofibrillar components of striated muscle. Proc Natl Acad Sci USA 76:3698–3702
Yamada A, Yoshio M, Kojima H, Oiwa K (2001) An in vitro assay reveals essential protein components for the “catch” state of invertebrate smooth muscle. Proc Natl Acad Sci USA 98:6635–6640. https://doi.org/10.1073/pnas.111585098
Yamasaki R et al (2001) Titin-actin interaction in mouse myocardium: passive tension modulation and its regulation by calcium/S100A1. Biophys J 81:2297–2313. https://doi.org/10.1016/S0006-3495(01)75876-6
Yarom R, Meiri U (1971) N lines in striated muscle: a site of intracellular Ca2+. Nat New Biol 234:254–256
Zhukarev V, Sanger JM, Sanger JW, Goldman YE, Shuman H (1997) Distribution and orientation of rhodamine-phalloidin bound to thin filaments in skeletal and cardiac myofibrils. Cell Motil Cytoskelet 37:363–377. https://doi.org/10.1002/(SICI)1097-0169(1997)37:4%3c363:AID-CM7%3e3.0.CO;2-5
Acknowledgements
We thank Stan Lindstedt for helpful comments on earlier versions of this manuscript. This work was supported by the National Science Foundation [IOS-0732949, IOS-1025806, IOS-1456868], the W. M. Keck Foundation, and the Technology Research Initiative Fund of Northern Arizona University.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nishikawa, K., Dutta, S., DuVall, M. et al. Calcium-dependent titin–thin filament interactions in muscle: observations and theory. J Muscle Res Cell Motil 41, 125–139 (2020). https://doi.org/10.1007/s10974-019-09540-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-019-09540-y