
Abstract
Muscle regeneration is a tightly orchestrated process where activated satellite cells (myoblasts) respond to external stimuli in order to proliferate, differentiate and fuse to damaged myofibers. Simultaneously, the injured tissue undergoes an inflammatory response and communication between leukocytes and the spectrum of differentiated and undifferentiated muscle cells is essential for proper healing. This communication is mediated by cytokines, growth factors and prostaglandins and dissecting the role of these signaling molecules might be the key to positively manipulate muscle regeneration in the future. This review will focus on the roles of prostaglandins and will consider the potential cost of using non-steroidal inflammatory drugs as popular treatment of muscle injury.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Skeletal muscle injuries arising following exercise, physical trauma or restricted blood supply are often debilitating and affect professional and day-to-day life. A detailed understanding of the healing process will help design therapies that promote quicker and better healing.
Muscle regeneration is a tightly orchestrated process where activated satellite cells (myoblasts) respond to external stimuli in order to proliferate, differentiate and fuse to damaged myofibers. Simultaneously, the injured tissue undergoes an inflammatory response and communication between leukocytes and the spectrum of differentiated and undifferentiated muscle cells is essential for proper healing. This communication is mediated by cytokines, growth factors and prostaglandins and dissecting the role of these signaling molecules might be the key to positively manipulate muscle regeneration in the future. This review will focus on the roles of prostaglandins (PGs) and will consider the potential cost of using non-steroidal inflammatory drugs (NSAIDs) as popular treatment of muscle injury.
The importance of inflammation in muscle healing
After injury, the damaged muscle tissue is quickly invaded by leukocytes that elicit an inflammatory response which is both beneficial and detrimental for healing (Tidball 2005). Initially, neutrophils release oxygen radicals and proteases to degrade cell debris generated during injury but, at the same time, cause further damage to the tissue (Tidball 2005). This is followed by the arrival of macrophages that remove cell debris by phagocytosis and secrete factors such as IGF, TGF-β, LIF, IL-6 and CSF-1 that modulate myoblast proliferation and differentiation (Cantini et al. 2002). Concurrently, satellite cells become active myoblasts that also secrete signaling molecules and, as inflammation resolves, commit to differentiation and fuse with the damaged myofibers. Thus, the rate and success of healing is influenced both positively and negatively by the cross-talk between myocytes and leukocytes.
Cyclooxygenases: the gateway for prostaglandin synthesis
Commonly, the immediate treatment for muscle injuries follows the ‘RICE’ principle (Rest, Ice, Compression and Elevation) in order to minimize the extent of the injury. In addition it is usual to recommend the use of NSAIDs to attenuate inflammation and pain (Jarvinen et al. 2007). NSAIDs exert their anti-inflammatory effects by inhibiting the COX enzymes therefore blocking prostaglandin synthesis (Funk 2001). Upon external stimulation (such as cytokines, growth factors or mechanical trauma) phospholipase A2 (PLA2) translocates from the cytoplasm to the perinuclear membranes where it catalyzes the release of arachidonic acid (AA) from phospholipids (Funk 2001) (Fig. 1). Then, through a two-step reaction, COXs convert AA into PGH2, the unstable precursor of all PGs. Finally, terminal synthases convert PGH2 into the five primary PGs: PGD2, PGE2, PGF2α, PGI2 (prostacyclin) or TXA2 (thromboxane) (Funk 2001; Simmons et al. 2004).

Prostaglandin synthesis pathway. Upon external stimulation (such as cytokines, growth factors or mechanical stress) phospholipase A2 (PLA2) translocates from the cytoplasm to the perinuclear membranes where it catalyzes the release of arachidonic acid (AA) from membrane phospholipids (PL). Cyclooxygenase (COX) enzymes quickly convert AA into prostaglandin (PG) H2, the unstable precursor of all PGs. COX activity is inhibited by non-steroidal anti-inflammatory drugs (NSAIDs). The fate of PGH2 is determined by the presence of the terminal PG synthases: PGE2 synthase (PGES), PGF2α synthase (PGFS), PGD2 synthase (PGDS), PGI2 synthase (PGIS) and thromboxane A2 synthase (TXAS). Furthermore, PGD2 spontaneously dehydrates to generate 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2). PGs are not stored in the cell but rather synthesized de novo upon stimulus and quickly released into the extracellular space where they interact with specific surface receptors
There are two COX isoforms with distinct physiological functions. COX-1 is constitutively expressed (often considered as “house-keeping gene”) that produces homeostatic PG levels. In contrast, COX-2 expression is generally repressed but can be induced by a variety of external stimuli causing a boost in PG production during pathophysiological processes such as inflammation and pain (Simmons et al. 2004). Traditional NSAIDs such as aspirin, ibuprofen and indomethacin inhibit both COX-1 and COX-2, but isoform-specific NSAIDs exist and are valuable tools to define the function of each enzyme in a biological system. Given the current clinical procedure for muscle injuries, it is essential to understand the role of COXs in muscle healing.
NSAID or not NSAID?
Many studies have attempted to determine the effect of COX inhibition during muscle regeneration using a wide variety of in vitro and in vivo models. Cyclical stretch increases proliferation in primary mouse myoblasts, accompanied by elevated PGE2 and PGF2α production. These effects are COX-2 dependent since myoblasts treated with SC-236 (COX-2 selective NSAID) and myoblasts derived from COX-2 null mice have no response to stretching (Otis et al. 2005). Whilst informative, in vitro models do not address physiological scenarios such as the role of inflammatory cells and their interaction with myoblasts. Mishra et al. subjected rabbits to exercise-induced injury and monitored the recovery after treatment with flurbiprofen (non-selective NSAID). Treated animals had better short-term recovery but a long-term deficit in force generation when compared to controls (Mishra et al. 1995). This observation highlights the concept that inflammation can be both beneficial and detrimental. Bondesen et al., focused on the role of COX-2 in muscle healing after injury (Bondesen et al. 2004) and atrophy (Bondesen et al. 2006). The expression of COX-2 (but not COX-1) increased after these challenges and ablation of enzyme activity with SC-236 or in COX-2 null mice resulted in reduced myofiber diameter upon healing. Importantly, these COX-2 deficient systems showed reduced myoblast and macrophage count in the regenerating environment confirming the importance of COX metabolites in the healing process. Altogether, these studies point to impaired healing after COX inhibition.
Muscle healing studies using human subjects are rare. Nevertheless recent publications have attempted to show the impact of NSAID intake after muscle injuries. Volunteers undertaking high-intensity resistance exercise showed an expected increase in muscle protein synthesis (Trappe et al. 2002). Also, PGE2 and PGF2α levels increased 64% and 77%, respectively (Trappe et al. 2001). However, intake of over-the-counter doses of ibuprofen suppressed protein and PG synthesis in response to exercise. In a follow-up study, the authors conclude that resistance exercise induces the expression of COX-2 but not COX-1 (Weinheimer et al. 2007). In another study, muscle biopsies from male volunteers were analyzed before and after a 36 km run. Eight days post-race, a 20% increase on satellite cell number was observed in the placebo group but not in the group taking a daily dose of 100 mg indomethacin (Mackey et al. 2007). Overall, these studies point to a crucial role of COX activity, particularly COX-2, in both myoblast and leukocyte intervention on muscle healing. However, it is important to keep in mind that the primary PGs downstream of COXs have distinct signaling properties and that the type of PGs produced during muscle healing might change across time.
What happens downstream of COX?
PGs are produced in nearly all cell types and signal in a paracrine or autocrine fashion. These short-lived molecules are not stored in the cell, but rather synthesized de novo from membrane lipids via the sequential actions of PLA2, COXs and PG terminal synthases (Funk 2001) (Fig. 1). PGs are quickly secreted to the extracellular space where they interact with specific high-affinity surface receptors, triggering an internal signal cascade (Funk 2001). In the regenerating muscle the expression of PG synthases, receptors and its downstream signaling pathways has only recently began to be described. In theory, modulating specific PG signaling in the healing muscle might be a more effective clinical approach than COX inhibition.
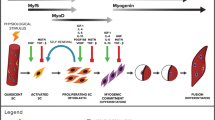
Proliferating myoblasts have consistently been shown to produce PGE2 and PGF2α (Bondesen et al. 2004; Otis et al. 2005). Early studies demonstrated that inhibition of myoblast fusion with indomethacin could be overcome by adding PGE2 (Entwistle et al. 1986). There are three known PGE synthases (PGES: mPGES-1, mPGES-2 and cPGES) and four PGE receptors (EP1, EP2, EP3 and EP4). However, the expression of these components of the PGE2 signaling pathway has not been characterized in the homeostatic and regenerating muscle. Thus the exact role of PGE2 in the healing context remains largely unknown. Interestingly, PGE2 is abundantly produced by leukocytes in the early phase of inflammation and could also affect myoblast differentiation (Fig. 2).

Prostaglandin (PG) signaling in the regenerating muscle. After muscle injury, satellite cells become proliferating myoblasts that later commit to differentiation and fuse to damaged myofibers. Simultaneously, leukocytes (mainly neutrophils and macrophages) invade the damaged tissue and initiate an inflammatory response. During this process both cell types secrete PGs that may influence myogenesis. Both myoblasts and leukocytes produce PGE2 a promoter of myogenesis, but no PGE synthases or receptors have been shown to be expressed in myoblasts. PGF2α is produced by myoblasts and signals via the PGF receptor (FP) to prevent apoptosis and promote cell fusion. Similarly, PGI2 (prostacyclin) signals in autocrine fashion via the PGI receptor (IP) to restrain myoblast motility and enhance cell fusion. PGD2 is produced by inflammatory leukocytes but its synthesis and signaling in myoblasts is unknown. However, 15d-PGJ2, a PGD2 dehydration metabolite, can inhibit myogenesis but the mechanism remains largely unknown
In isolated muscles PGF2α was described as a promoter of protein synthesis and its production is increased upon exercise (Trappe et al. 2001). PGF2α signals via one receptor, FP, resulting in elevation of intracellular calcium (Funk 2001). Horsley et al., reported that primary mouse myoblasts treated with PGF2α generated myotubes of increased size by stimulating fusion with nascent fibers (Horsley and Pavlath 2003). This growth was FP dependent and caused the activation of the calcium-regulated nuclear factor of activated T cells, isoform C2 (NFATC2). In a follow-up study, the authors show that PGF2α increases myotube size by preventing myoblast apoptosis, via the upregulation of BIR ubiquitin-conjugating enzyme (BRUCE), an inhibitor of caspases (Jansen and Pavlath 2008). BRUCE was naturally expressed in the later phases of adult myogenesis and its upregulation was ablated in NFATC2 null myoblasts. Importantly, PGF2α treated or BRUCE over-expressing myoblasts engraft more efficiently on a transplant model. These two studies characterized in detail the autocrine signaling pathway of PGF2α in myoblasts, confirming an important myogenic role of a COX product.
PGI2 (also known as prostacyclin) is the main COX metabolite produced by vascular endothelial cells where it promotes vasodilation. Primary mouse myoblasts were shown to express PGI synthase, PGI receptor (IP) and produce PGI2 (Bondesen et al. 2007). IP null myoblasts had reduced fusion and differentiation but increased cell motility. The high motility rate of IP null myoblasts was blocked by Iloprost (stable PGI2 analog) resulting in increased cell fusion. The authors propose that myoblast motility is inversely correlated with differentiation and that PGI2 plays a pivotal role in this balance.
PGD2: the forgotten prostaglandin in muscle repair?
Studies in other cell systems have shown that like PGE2, PGD2 synthesis is strongly induced in the early phase of inflammation (Rajakariar et al. 2007) and can have both pro- and anti-inflammatory roles, depending on the cell-type context. In spite of the importance of inflammation in muscle healing, PGD2 synthesis and signaling has not been characterized in myoblasts. This could perhaps unravel a novel cross-talk pathway between inflammatory and muscle cells.
In solution, PGD2 undergoes spontaneous dehydration to generate 15-deoxy-Δ12,14-PGJ2 (15d-PGJ2) (Fig. 1), a potent electrophilic PG capable of generating reactive oxygen species, covalently modifying cellular proteins such as NF-κB and activating the nuclear receptor peroxisome proliferator-activated receptor γ (PPARγ) (Uchida and Shibata 2008). Like PGD2, levels of 15d-PGJ2 are increased during inflammation (Rajakariar et al. 2007) and might modulate myoblast behavior. Although PPARγ expression seems to be important in C2C12 myoblast differentiation (Singh et al. 2007), it was observed that 15d-PGJ2 blocked myotube formation in a PPARγ-independent manner (Hunter et al. 2001). This differentiation block could be mediated by the other properties of 15d-PGJ2 such as the generation of an oxidative environment which has been shown to impair myogenesis (Ardite et al. 2004). Also, 15d-PGJ2 has been shown to cross-react with other PG receptors (Hata et al. 2003).
Conclusion
Data from animal and human studies supports that COX activity is important in muscle healing. It is now essential to understand how individual PGs act downstream of COX having in mind the sequential profile of inflammation and myogenesis (Fig. 2). Muscle injury induces COX-2 dependent secretion of PGE2, PGI2 and PGF2α by activated myoblasts. The mechanism of signaling by PGI2 and PGF2α has been well described but much is yet to be known about PGE2. Simultaneously, invading leukocytes are capable of producing PGE2, PGD2 and 15d-PGJ2, whose functions have been well described in inflammation. However, the impact of such leukocyte-derived PGs on myogenesis is poorly understood. Isolated primary myoblast studies tend to focus on endogenously produced PGs thus ignoring possible roles of leukocyte PG-mediated communication. Co-culture studies analyzing the interaction between myoblasts and leukocytes might bring some answers to the topic of cellular cross-talk in muscle healing. A complete profiling of all PG synthases and receptors in different stages of adult myogenesis would inform which PG pathways might be active at each stage. Eventually, with the development of drugs that specifically modulate PG synthases and receptors, it could be possible to clinically improve the current therapies for muscle injury.
References
Ardite E, Barbera JA, Roca J, Fernandez-Checa JC (2004) Glutathione depletion impairs myogenic differentiation of murine skeletal muscle C2C12 cells through sustained NF-kappaB activation. Am J Pathol 165:719–728
Bondesen BA, Mills ST, Kegley KM, Pavlath GK (2004) The COX-2 pathway is essential during early stages of skeletal muscle regeneration. Am J Physiol Cell Physiol 287:C475–C483. doi:10.1152/ajpcell.00088.2004
Bondesen BA, Mills ST, Pavlath GK (2006) The COX-2 pathway regulates growth of atrophied muscle via multiple mechanisms. Am J Physiol Cell Physiol 290:C1651–C1659. doi:10.1152/ajpcell.00518.2005
Bondesen BA, Jones KA, Glasgow WC, Pavlath GK (2007) Inhibition of myoblast migration by prostacyclin is associated with enhanced cell fusion. FASEB J 21:3338–3345. doi:10.1096/fj.06-7070com
Cantini M, Giurisato E, Radu C, Tiozzo S, Pampinella F, Senigaglia D, Zaniolo G, Mazzoleni F, Vitiello L (2002) Macrophage-secreted myogenic factors: a promising tool for greatly enhancing the proliferative capacity of myoblasts in vitro and in vivo. Neurol Sci 23:189–194. doi:10.1007/s100720200060
Entwistle A, Curtis DH, Zalin RJ (1986) Myoblast fusion is regulated by a prostanoid of the one series independently of a rise in cyclic AMP. J Cell Biol 103:857–866. doi:10.1083/jcb.103.3.857
Funk CD (2001) Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294:1871–1875. doi:10.1126/science.294.5548.1871
Hata AN, Zent R, Breyer MD, Breyer RM (2003) Expression and molecular pharmacology of the mouse CRTH2 receptor. J Pharmacol Exp Ther 306:463–470. doi:10.1124/jpet.103.050955
Horsley V, Pavlath GK (2003) Prostaglandin F2(alpha) stimulates growth of skeletal muscle cells via an NFATC2-dependent pathway. J Cell Biol 161:111–118. doi:10.1083/jcb.200208085
Hunter JG, van Delft MF, Rachubinski RA, Capone JP (2001) Peroxisome proliferator-activated receptor gamma ligands differentially modulate muscle cell differentiation and myod gene expression via peroxisome proliferator-activated receptor gamma-dependent and -independent pathways. J Biol Chem 276:38297–38306
Jansen KM, Pavlath GK (2008) Prostaglandin F2alpha promotes muscle cell survival and growth through upregulation of the inhibitor of apoptosis protein BRUCE. Cell Death Differ
Jarvinen TA, Jarvinen TL, Kaariainen M, Aarimaa V, Vaittinen S, Kalimo H, Jarvinen M (2007) Muscle injuries: optimising recovery. Best Pract Res Clin Rheumatol 21:317–331. doi:10.1016/j.berh.2006.12.004
Mackey AL, Kjaer M, Dandanell S, Mikkelsen KH, Holm L, Dossing S, Kadi F, Koskinen SO, Jensen CH, Schroder HD, Langberg H (2007) The influence of anti-inflammatory medication on exercise-induced myogenic precursor cell responses in humans. J Appl Physiol 103:425–431. doi:10.1152/japplphysiol.00157.2007
Mishra DK, Friden J, Schmitz MC, Lieber RL (1995) Anti-inflammatory medication after muscle injury. A treatment resulting in short-term improvement but subsequent loss of muscle function. J Bone Joint Surg Am 77:1510–1519
Otis JS, Burkholder TJ, Pavlath GK (2005) Stretch-induced myoblast proliferation is dependent on the COX2 pathway. Exp Cell Res 310:417–425. doi:10.1016/j.yexcr.2005.08.009
Rajakariar R, Hilliard M, Lawrence T, Trivedi S, Colville-Nash P, Bellingan G, Fitzgerald D, Yaqoob MM, Gilroy DW (2007) Hematopoietic prostaglandin D2 synthase controls the onset and resolution of acute inflammation through PGD2 and 15-deoxyDelta12 14 PGJ2. Proc Natl Acad Sci USA 104:20979–20984. doi:10.1073/pnas.0707394104
Simmons DL, Botting RM, Hla T (2004) Cyclooxygenase isozymes: the biology of prostaglandin synthesis and inhibition. Pharmacol Rev 56:387–437. doi:10.1124/pr.56.3.3
Singh J, Verma NK, Kansagra SM, Kate BN, Dey CS (2007) Altered PPAR gamma expression inhibits myogenic differentiation in C2C12 skeletal muscle cells. Mol Cell Biochem 294:163–171. doi:10.1007/s11010-006-9256-x
Tidball JG (2005) Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol 288:R345–R353. doi:10.1152/ajpregu.00454.2004
Trappe TA, Fluckey JD, White F, Lambert CP, Evans WJ (2001) Skeletal muscle PGF(2)(alpha) and PGE(2) in response to eccentric resistance exercise: influence of ibuprofen acetaminophen. J Clin Endocrinol Metab 86:5067–5070. doi:10.1210/jc.86.10.5067
Trappe TA, White F, Lambert CP, Cesar D, Hellerstein M, Evans WJ (2002) Effect of ibuprofen and acetaminophen on postexercise muscle protein synthesis. Am J Physiol Endocrinol Metab 282:E551–E556
Uchida K, Shibata T (2008) 15-Deoxy-Delta(12, 14)-prostaglandin J2: an electrophilic trigger of cellular responses. Chem Res Toxicol 21:138–144. doi:10.1021/tx700177j
Weinheimer EM, Jemiolo B, Carroll CC, Harber MP, Haus JM, Burd NA, LeMoine JK, Trappe SW, Trappe TA (2007) Resistance exercise and cyclooxygenase (COX) expression in human skeletal muscle: implications for COX-inhibiting drugs and protein synthesis. Am J Physiol Regul Integr Comp Physiol 292:R2241–R2248. doi:10.1152/ajpregu.00718.2006
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Veliça, P., Bunce, C.M. Prostaglandins in muscle regeneration. J Muscle Res Cell Motil 29, 163–167 (2008). https://doi.org/10.1007/s10974-008-9154-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10974-008-9154-9