
Abstract
Betulonic acid (BA) is a pentacyclic lupane-type triterpenoid possessing valuable pharmacological activities, exhibiting very low water solubility. Inclusion complexes between the substance and cyclodextrins (CDs) were obtained in order to improve its solubility and consequently its bioavailability. The aim of this study was to investigate the guest–host interaction of BA with γ-cyclodextrin (γ-CD) and its derivative, 2-hydroxypropyl-γ-cyclodextrin (HP-γ-CD), in solution and in solid state in order to prove the formation of inclusion complexes between the components. The kneading method was used for the inclusion complexes preparation, and different analytical techniques such as thermal analysis, powder X-ray diffractometry, universal attenuated total reflectance Fourier transform IR spectroscopy (UATR-FTIR) and UV spectroscopy were employed to investigate the interaction between substances. The stoichiometry of the inclusion complex BA/HP-γ-CD was found to be 1:1 by employing continuous variation method, and the apparent stability constant was calculated as 1855.55 M−1 using Benesi–Hildebrand equation. Structural studies of the inclusion complexes were carried out using molecular modeling techniques in order to explain the complexation mechanism. The results of this study confirm the formation of inclusion complexes between BA and cyclodextrins both in solution and in solid state.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Natural products have always been an important source of starting compounds for drug discovery and development [1,2,3]. They have played a substantial role in the antitumor drug development [4, 5], a great majority of antitumor compounds originate in natural products [4, 6,7,8]. The biological active compounds contained in natural products have special selectivity to cellular targets [3, 9].
Pentacyclic triterpenoids are one of the most abundant natural compounds in the plant kingdom [4, 8] and, in the last decade, have received particular attention due to their important medicinal properties [10,11,12]. Betulonic acid (BA, 3-oxolup-20(29)-en-28-oic acid, Fig. 1) is a triterpenoid with lupane skeleton, presenting poor water solubility (in silico predicted aqueous solubility − 8.0 < log(Sw) < − 6.0) [13]. BA and its derivatives have proved biological activities including antiviral [14,15,16], anti-inflamatory [17, 18], antitumor [4, 19], hepatoprotective [18, 20], anti-HCMV activity [21], antimicrobial [22], immunostimulant [23] and antioxidant effects [24].

Chemical structure of betulonic acid (BA) (a) and of γ-cyclodextrin, with the location attachment of the hydroxypropyl group (b) or R-hydroxypropyl (c)
Cyclodextrins (CDs) belong to the family of the cyclic oligosaccharides and possess six, seven and eight glucopyranose units linked by α-(1,4) bonds (α, β and γ-cyclodextrin, respectively). CDs molecules have torus macro-ring shape, like a truncated cone, having a hydrophobic internal cavity and a hydrophilic outer surface, responsible for their water solubility [25,26,27]. This particular structure of CDs offers them the ability to form host-guest inclusion complexes with a wide variety of drug substances of appropriate shape and size. The inclusion complex formation is of biomedical and pharmaceutical interest because of the major changes in drug candidate physicochemical and biopharmaceutical properties, such as solubility, chemical reactivity, stability, volatility, melting point, unpleasant taste or odors, which lead to significant amelioration of formulation design and improvement of drugs bioavailability [27,28,29,30,31,32,33]. Additionally, CDs can also reduce ocular and gastrointestinal irritation and prevent drug-excipient and drug-drug interactions [25, 29, 34]. Thus, the CD complexation results in significant improvement of drugs formulation design and enhancement of solubility, dissolution and bioavailability of drugs [25, 35,36,37].
Some studies have reported the inclusion complexes formation between pentacyclic triterpenoids such as betulinic, oleanolic and ursolic acid, betulin and cyclodextrins, including β, γ-cyclodextrin and its derivatives [38,39,40,41,42,43,44].
In order to overcome the very low water solubility of betulonic acid, Saxena B et al. [19] have synthesized a hydrophilic lysinated derivative of drug with anti-prostate cancer activity. Up to the present, there has been no scientific report on BA inclusion complex with γ-CD and its derivatives. Following this, the aim of this paper has been to investigate the molecular encapsulation of BA in γ-cyclodextrin and 2-hydroxypropyl-γ-cyclodextrin cavity. The binary products have been prepared using the kneading method in molar ratio 1:1. Physicochemical determinations based on thermal analyses, PXRD, FTIR and UV spectroscopy have been used to characterize the inclusion complexes both in solid state and in solution.
Experimental
Materials
Betulin, as starting material, was obtained in our laboratory by Soxhet extraction from the white outer birch bark (Betula pendula) [45]. The functionalizated method for the BA obtaining was previously described by our group [45]: Betulin was oxidized with freshly prepared Jones’ reagent (consisting in dissolved CrO3 in a mixture of concentrated H2SO4 and water) in acetone. After purification procedure, BA heaving purity > 96% was obtained. γ-CD and HP-γ-CD were purchased from Cyclolab R&L Ltd (Budapest, Hungary). The substances were used as received. All other chemicals and reagents were of analytical grade.
Characterization of inclusion complexes in solution
All spectrophotometric measurements were recorded using SPECTRONIC UNICAM—UV 300 UV-VISIBLE double beam spectrophotometer with 1 cm matched quartz cells.
Stoichiometry and stability constant determination
The stoichiometry of the BA/γ-CD and BA/HP-γ-CD inclusion complexes was investigated by Job’s method [29, 46]. Job plot was obtained from UV spectrophotometric measurements. For this purpose, equimolar 2.2 × 10−4 M solution of BA and CDs (γ-CD and HP-γ-CD, respectively) prepared in 0.1 M phosphate buffer (pH 8.5) was mixed to a standard volume, varying the molar ratio, but keeping the total concentration of the species constant. An analogous dilution set of the BA stock solution was prepared using the same solvent. After stirring, the absorbance was measured at 205 nm for all solution, and the difference in absorbance ΔA (ΔA = A0 – A) of BA in the presence (A) and in the absence (A0) of CDs was plotted against BA mole fraction R; R = [BA]/{[BA] + [CD]}.
The apparent stability constant of the BA/HP-γ-CD inclusion complex was determined employing Benesi–Hildebrand model. The spectrophotometric data were collected for BA in the presence of increasing concentration of CD and also under free complexation conditions. The BA concentration was maintained constant, at 1.759 × 10−4 M in 0.1 M phosphate buffer (pH 8.5), and the CD concentration was varied from 0 to 1.759 × 10−3 M, in the same solvent. The absorption spectra were recorded in the range of 200–230 nm, using 1.0 cm quartz cells.
The binding constant determination is achieved by using the changes in absorbance at maximum wavelength of 205 for BA in the presence of increasing CD concentration. The stability constant value for the 1:1 inclusion complex can be obtained by means of the Benesi–Hildebrand equation [29, 46, 47]
where Δε is change in molar attenuation coefficient, ΔA is change in absorbance and K is stability constant.
Preparation of solid inclusion complexes BA/γ-CD and BA/HP-γ-CD
The kneading method in 1:1 molar ratio was used to prepare the inclusion complexes. This method was applied because of its simplicity, accessibility, rapidity and economical advantages in comparison with other methods [48]. The amounts of 0.0389 g BA and 0.1111 g γ-CD were weighed, and the obtained mixture was pulverized in a mortar and triturated with 0.15 g ethanol-water solution (1:1, m/m). In the case of BA/HP-γ-CD inclusion complex preparation, 0.0378 g BA and 0.1119 g HP-γ-CD were weighed and the obtained mixture was pulverized and triturated with 0.15 g ethanol-water solution (1:1, m/m) in order to get a homogeneous paste. The paste thus obtained was kneaded for 45 min, and during this process, a few drops of solvent were added in order to ensure a suitable consistency. The product was dried at room temperature and then in the oven, at 40 °C for 24 h. The dried kneaded products named BA/γ-CD and BA/HP-γ-CD were pulverized and passed through a 75 µm size sieve.
Characterization of solid-state inclusion complexes
Thermal analysis
TG/DTG/HF measurements were performed using a Perkin Elmer DIAMOND TG/DTA instrument. In these experiments, the samples of about 3–4 mg were placed in the aluminum crucibles. The thermal behavior of BA, γ-CD, HP-γ-CD, the binary systems BA/γ-CD and BA/HP-γ-CD has been studied under air atmosphere at a flow rate of 100 mL min−1, in the temperature range of 40–500 °C, with heating rate of 10 °C min−1.
Powder X-ray diffractometry
X-ray diffraction patterns of BA, γ-CD, HP-γ-CD and of the binary systems BA/γ-CD and BA/HP-γ-CD were obtained using a Bruker D8 Advance powder X-ray diffractometer, at ambient temperature, in the range of 10°–45° angular domain (2θ). The samples were irradiated with Ni-filtered CuKα radiation (40 kV, 40 mA).
Fourier transform infrared spectroscopy
The FTIR spectra were recorded using a Perkin Elmer SPECTRUM 100 device. The data were collected directly on solid samples, in the spectral domain 4000–600 cm−1 on an UATR device. Spectra were built up after a number of 16 coadded acquisitions, with a spectral resolution of 4 cm−1.
Molecular modeling
The Gaussian program suite at DFT/B3LYP/6-311G for BA optimization was used. The γ-CD X-ray crystal structure (with 5E70 code and X-ray diffraction at 2.33 Å resolution) was taken from the Protein Data Bank [49] and then optimized with the Gaussian software Hartree–Fock/6-311G. The HP-γ-CD molecular structure was modeled by adding eight hydroxypropyl moieties to the γ-CD structure, attached to the primary hydroxyl groups from carbon six D-glycopyranosyl units, then optimized in the same manner (DFT/B3LYP/6-311G). The molecular docking analysis was performed using the AutoDock 4.2.6 software together with the AutoDockTools, (a molecular viewer and graphical support for setup and analysis of docking runs). The preparation of γ-CD host molecule involves adding all the hydrogens, computing the Gasteiger charge; the grid box was created using Autogrid 4 with 50 × 40 × 40 Å in x, y and z directions with 0.375 Å spacing. All the calculations were performed in vacuum.
For the docking process, we chose the Lamarckian genetic algorithm (Genetic Algorithm combined with a local search), with a population size of 150 and a number of 30 runs. We exported all AutoDock results in the PyMOL molecular visualization system [50].
Results and discussion
Characterization of inclusion complexes in solution
Stoichiometry and stability constant determination
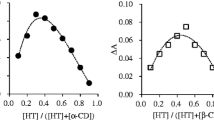
According to the continuous variation method [51], the stoichiometry of the inclusion complex is given by the value of the molar ratio R that corresponds to the maximum concentration of the complex. The molar ratio is identified when the absorbance (a physical parameter which is directly related to the complex concentration) was measured for a series of samples having continuously varied molar fraction of its components [29, 46, 52]. The maximum absorbance variation (ΔA) for BA, as shown in Fig. 2a, is reached, when BA mole fraction is 0.667 in the presence of γ-CD, which may indicate that the main stoichiometry of BA/γ-CD inclusion complex is 2:1. In the case of BA/HP-γ-CD inclusion complex formation, the maximum variation in absorption is observed for R = 0.5, thus indicating that the stoichiometry is 1:1 (Fig. 2b).

Job’s plot for BA/γ-CD (a) and BA/HP-γ-CD (b) inclusion complexes from absorbance measurements
The Benesi–Hildebrand method is one of the common strategies for estimation of the stoichiometry and binding constant of the inclusion complexes [29, 46, 47]. For the purpose of confirming the 1:1 stoichiometry of the BA/HP-γ-CD inclusion complex and calculating its stability constant, changes in the absorption intensity of BA at 205 nm have been evaluated as a function of HP-γ-CD concentration. A hypochromic shift is observed in the absorption maxima of BA in the presence of HP-γ-CD compared with pure BA, supporting the hypothesis of an inclusion complex formation. Also, from Fig. 3a it can be seen an increase of BA absorption intensity as a consequence of increasing HP-γ-CD concentrations. The variation of the absorbance ΔA = A − A0 was calculated as the difference between BA absorbance in the presence of different HP-γ-CD concentrations and the pure BA absorbance.

UV spectra of pure BA and BA/HP-γ-CD inclusion complex in presence of increasing HP-γ-CD concentration (a); Benesi–Hildebrand linear plot for 1/ΔA versus 1/[HP-γ-CD] (b)
For the aim of verifying the inclusion complex stoichiometry, a double reciprocal plot was represented, 1/ΔA versus 1/[HP-γ-CD], as it is shown in Fig. 3b. A good linear relationship was achieved when 1/ΔA was plotted against 1/[HP-γ-CD], (R2 = 0.9884), confirming the 1:1 stoichiometry of the BA/HP-γ-CD inclusion complex.
The value of the apparent stability constant was obtained using Benesi–Hildebrand Eq. (1), as a ratio of the intercept to slope of the straight line in the Benesi–Hildebrand double reciprocal plot. The stability constant value was determined as K1:1 = 1855.55 M−1.
Characterization of solid-state inclusion complexes
Thermal analysis
In order to evaluate the interaction between BA and CDs, the thermal behavior of the pure substances and their kneaded products was investigated using TG, DTG and HF.
TG, DTG and HF thermoanalytical curves of BA, γ-CD, HP-γ-CD and their inclusion complexes are shown in Fig. 4a–e.

The thermal profile of: BA (a); γ-CD (b); BA/γ-CD kneaded product (c); HP-γ-CD (d) and BA/HP-γ-CD (e) binary system in air atmosphere (100 mL min−1), temperature range of 40–500 °C and heating rate of 10 °C min−1
The TG/DTG curves of BA reveal a multistage decomposition process in air atmosphere (Fig. 4a). A good thermal stability of the substance is noticed, the fist mass loss starting up at 222 °C with a DTGmax = 273 °C (Δm = 12%), followed by another process which begins at 286.7 °C, DTGmax = 307.6 °C (Δm = 41%). These two mass loss steps are accompanied by a strong exothermic effect in HF curve (Tpeak = 310 °C), which probably corresponds to the thermo-oxidations of the triterpenic moiety. The BA heating at 525 °C induces a total mass loss of 100% [53].
The thermoanalytical curves of γ-CD present a small mass loss between 40 and 130 °C (Δm = 9.4%) corresponding to the loss of crystal water (Fig. 4b); a stability stage of dehydrated γ-CD is noticed between 130–260 °C, but after 260 °C the mass loss process continues. The γ-CD melting occurs at 318.6 °C, as the endothermic peak of the HF curve shows.
The TG/DTG curves of HP-γ-CD indicate a mass loss up to 100 °C (Δm = 8.4%) due to the cyclodextrin dehydration (Fig. 4d), followed by a thermal stability stage in the temperature range of 100–265 °C. The HF curve reveals the HP-γ-CD melting at 334.5 °C.
The thermal profiles of the binary systems reveal differences in comparison with those of the pure substances (Fig. 4c, e). The thermoanalytical curves of BA/γ-CD kneaded product present a mass loss process between 40 and 95 °C (Δm = 8.2%) corresponding to γ-CD dehydration; in the temperature range of 95–225 °C a stability stage is observed, but after 225 °C the mass loss process continues. It is also noticed a marked reduction in intensity of the DTG peak corresponding to the BA first mass loss step in the range 200–400 °C and a shift toward higher temperature (DTGmax = 285 °C). The HF curve of the binary system BA/γ-CD indicates the significant reduction and the displacement of both melting endothermic peak of γ-CD (i.e. 308 °C) and the BA thermo-oxidations exothermic peak (i.e., 328.4 °C), as compared with the parent substances.
Furthermore, the endothermic peak corresponding to the HP-γ-CD melting exhibits a marked reduction in its intensity and shifts to lower temperature (i.e., 322.6 °C) in the HF curve of the BA/HP-γ-CD kneaded product and also, the exothermic peak due to the thermo-oxidations of the BA triterpenic moiety is shifted to higher temperature (i.e., 331.6 °C).
The thermal analysis represents a valuable technique regarding the evaluation of the interactions between CDs and drug substances. The melting point of the guest molecules which are embedded in CDs cavity generally shifts to a different temperature and decreases its intensity or disappears [54, 55]. The area reduction and the shift toward a higher temperature of the thermo-oxidations of BA or shift toward a lower temperature of the melting peaks of CDs in the kneaded products indicate a molecular interaction between the guest and host molecules.
The results obtained by using the thermal analysis indicate a molecular interaction between the BA and CDs, through the inclusion complexes formation when the kneading method was employed.
Powder X-ray diffractometry
The powder X-Ray diffractograms of BA, γ-CD, HP-γ-CD and BA/γ-CD, respectively, BA/HP-γ-CD inclusion complexes are depicted in Fig. 5.

XRD patterns of BA, γ-CD, BA/γ-CD kneaded product (a) and BA, HP-γ-CD, BA/HP-γ-CD binary system (b)
The XRD pattern of BA had one broad peak and many undefined, diffused peaks with low intensities, reflecting the amorphous nature of substance, while the diffraction pattern of γ-CD contains a number of sharp and intense peaks at 2θ 12.03; 13.64; 15.13; 16.23; 18.42, reflecting its crystalline nature. The binary system BA/γ-CD diffractometric profile presents different degrees of changes in comparison with those of the pure substances. Thus, the characteristic diffraction peaks of γ-CD have disappeared in the XRD pattern of kneaded product, while there can be noticed the appearance of some broad peaks indicating that a new amorphous solid phase is formed.
The diffraction pattern of HP-γ-CD indicates its amorphous structure. The binary system BA/HP-γ-CD diffractogram emphasizes its amorphous nature, different from that of BA, by the two broad peaks and many undefined, diffused peaks with low intensities, similar to that of pure cyclodextrin.
The XRD analysis suggests that new compounds with an amorphous state have probably been formed.
UATR-FTIR analysis
The UATR-FTIR spectra of pure BA, γ-CD, HP-γ-CD and the BA binary products with CDs are presented in Fig. 6. The IR spectra of the kneaded products have been evaluated as compared to those of the pure substances in order to assess the inclusion complexes formation.

UATR-FTIR spectra of BA, γ-CD and their inclusion complex (a); BA, HP-γ-CD and their kneaded product (b)
The UATR-FTIR spectral pattern of BA shows a strong band at 1698 cm−1 associated to the C=O stretching vibration of ketone and carboxyl group. It also presents other characteristic bands for the carboxyl group (the C–O stretching at 1180 cm−1, the hydrogen bonded O-H stretch vibration at 3442 cm−1), for the vinylidene group (the C=C stretching vibration at 1642 cm−1 [45], the C–H stretch vibration at 3081 cm−1) and for the lupan skeleton (the sp3 C-H stretching vibration from CH3 and cycle CH2 at 2947 and 2867 cm−1, the CH2 scissoring at 1459 cm−1 and the methyl C-H antisymmetric/symmetric bending at 1459 cm−1 and 1376 cm−1, respectively) [45, 56]. The band located at 888 cm−1 can be assigned to either the C-H out-of-plane bending from vinylidene group or the antisymmetric cyclohexane ring stretching vibration [56, 57].
The FTIR spectra of the CDs indicate a broad band between 3600 and 3000 cm−1 associated to the O-H stretching vibration and characteristic peaks that correspond to the C-H stretching vibration from CH2 (at 2927 cm−1 for γ-CD; 2926 cm−1 for HP-γ-CD), to C–H bending from CH2 (1365 cm−1 for γ-CD; 1366 cm−1 for HP-γ-CD), to the C–C–O stretching vibration from C–OH (1021 cm−1 for γ-CD; 1014 cm−1 for HP-γ-CD) and to the skeletal vibration involving α 1-4 linkage (at 935 cm−1 for both γ-CD and HP-γ-CD). These data are in agreement with previously reported results [58].
The IR spectra of the kneaded products reveal major differences in comparison with the corresponding parent substances. As can be seen, almost all BA characteristic peaks have either reduced their intensity and shift to other wavenumbers or have disappeared in the IR spectrum of the binary compounds. Spectral data indicate that the BA characteristic bands corresponding to the carboxyl group, vinylidene group and the BA bands from 1376 and 888 cm−1 are present neither in BA/γ-CD kneaded product nor in BA/HP-γ-CD binary system. Furthermore, the BA CH2 scissoring band is still present in the BA/HP-γ-CD kneaded product spectrum and shifted to 1452 cm−1, but is no more present in the spectrum of the BA/γ-CD binary product and the BA band assigned to C-H stretching vibration from CH3 shifted from 2947 cm−1 in the pure compound to 2940 cm−1 in both inclusion complexes spectra, as a consequence of BA-CDs interaction when the kneading method was employed. Also, the bands assigned to C-H stretching vibration in CDs spectra shifted to 2940 cm−1 in spectra of inclusion complexes.
The results of the spectral analysis reveal changes in the characteristic bands of the pure compounds, thus indicating an interaction between the BA and CDs through the inclusion complexes formation.
Molecular modeling
Molecular modeling is a powerful tool that theoretical chemistry employs for quantitative predictions on host-guest interaction [52].
AutoDock(1) is an automated procedure for predicting the interaction of ligands with their macromolecular targets. An ideal procedure should be able to find the global minimum in the interaction energy between the substrate and the target protein, exploring all available degrees of freedom for the system. AutoDock thus combines two methods to achieve these goals: rapid grid-based energy evaluation and efficient search of torsional space. The conformations are then evaluated using a semi-empirical free energy force field. The force field includes six pair-wise evaluations (V) and an estimate of the conformational entropy lost upon binding \(( \otimes S_{\text{conf}} )\):
where L refers to the “ligand” and P refers to the “protein” in a ligand-protein docking calculation. Each of the pair-wise energetic terms includes evaluations for dispersion/repulsion, hydrogen bonding, electrostatics and desolvation:
The weighting constants W have been optimized in order to calibrate the empirical free energy based on a set of experimentally determined binding constants. The first term is a 6/12 potential for dispersion/repulsion interactions. The parameters are based on the Amber force field. The second term is a directional H-bond term based on a 10/12 potential. The parameters C and D are assigned to give a maximal well depth of 5 kcal mol−1 at 1.9 Å for hydrogen bonds with oxygen and nitrogen, and a well depth of 1 kcal mol−1 at 2.5 Å for hydrogen bonds with sulfur. The function E(t) provides directionality based on the angle t from ideal H-bonding geometry. The third term is a screened Coulomb potential for electrostatics. The final term is a desolvation potential based on the volume of atoms (V) that surround a given atom and shelter it from solve nt, weighted by a solvation parameter (S) and an exponential term with distance-weighting factor ∫ = 3.5 Å [59].
The free energy of binding that we will attempt to estimate is calculated as ΔbindG = ΔH − TΔS, where the ΔH represents the enthalpic, and ΔS the entropic contribution (only a negative ΔG value is energetically favorable and the process is considered spontaneous).
The binding free energy values are calculated as follows: − 8.90 kcal mol−1 for BA/γ-CD inclusion complex, − 5.29 kcal mol−1 for the 2 BA/γ-CD inclusion complex and − 5.95 kcal mol−1 for the BA/HP-γ-CD. The negative binding free energy values hint that the inclusion complexation process is favorably driven by enthalpy, thus confirming the interaction between the host molecules, namely γ-CD and HP-γ-CD, and the guest BA molecules. According to our data, the BA/HP-γ-CD inclusion complex is more stable than the 2 BA/γ-CD, because it has a lower value of binding free energy. Figures 7 and 8 present the theoretical BA/γ-CD and BA/HP-γ-CD inclusion complexes, as rendered in the PyMOL molecular visualization system. Figure 7 shows the molecular modeled inclusion complex BA/γ-CD, simulated in 2:1 molar ratio.

Inclusion complex simulation for a 2:1 molar ratio between BA and γ-CD. Images (a) and (b) show the inclusion complex from the narrow rim of γ-CD’s cavity. Images in (c) and (d) are lateral views that indicate the BA guest molecule entering within γ-CD through the wider cavity rim. BA host molecules are represented in blue, while γ-CD is represented in purple

PyMOL visualization of the BA/HP-γ-CD inclusion complex. Images (a–d) are lateral views of the inclusion complex. The BA guest molecules are shown in blue, while HP-γ-CD is represented in green
Figure 8 presents the molecular modeling images of the BA/HP-γ-CD inclusion complex.
Our molecular docking results bring out a structural insight about BA interactions with both γ-CD and HP-γ-CD, which supports the experimental results that indicate the formation of inclusion complexes between the BA and the two host cyclodextrins. However, in our future studies, we will investigate also the influence of complexation over decomposition mechanism of inclusion complexes, by already established protocols in our research group [60,61,62,63,64].
Conclusions
In this study, the inclusion complexes of BA and two CDs, γ-CD and HP-γ-CD were prepared and evaluated by experimental and theoretical approaches. Job’s method indicated a 1:1 stoichiometry for the BA/HP-γ-CD inclusion complex and 2:1 stoichiometry in the case of BA/γ-CD inclusion complex. The results obtained for the solid inclusion complexes evaluation, using techniques such as the thermal analysis, UATR-FTIR spectroscopy and PXRD clearly demonstrate that the binary products possess different physicochemical properties in relation to the pure compounds. The above results indicate that the kneading method leads to formation of solid-state complexes between BA and the two cyclodextrins.
References
Gu J, Gui Y, Chen L, Yuan G, Lu H-Z, Xu X. Use of natural products as chemical library for drug discovery and network pharmacology. PLoS ONE. 2013;8(4):e62839. https://doi.org/10.1371/journal.pone.0062839.
Newman DJ, Cragg G. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–35.
Ledeţi I, Avram Ş, Bercean V, Vlase G, Vlase T, Ledeţi A, Zupko I, Mioc M, Şuta LM, Şoica C, Dehelean C. Solid-state characterization and biological activity of betulonic acid derivatives. Molecule. 2015;20(12):22691–702.
Yang SJ, Liu MC, Zhao Q, Hu DY, Xue W, Yang S. Sinthesis and biological evaluation of betulonic acid derivatives as antitumor agents. Eur J Med Chem. 2015;96:58–65.
Dev S. Impact of natural products in modern drug development. Indian J Exp Biol. 2010;48(3):191–8.
Shoeb M. Anticancer agents from medicinal plants. Bangl J. Pharmacol. 2006;1:35–41.
Newman DJ, Gragg GM, Snader KM. The influence of natural products upon drug discovery. Nat Prod Rep. 2000;17:215–34.
Domingues RMA, Oliveir ELG, Freire CSR, Couto RM, Simoes PC, Neto CP, Silvestre AJD, Silva CM. Supercritical fluid extraction of Eucalyptus globules bark-A promising approach for triterpenoid production. Int J Mol Sci. 2012;13:7648–62.
Lagunin A, Filimonov D, Poroikov V. Multi-targeted natural products evaluation based on biological activity prediction with PASS. Curr Pharm Des. 2010;16:1703–17.
Jager S, Trojan H, Kopp T, Laszczyk MN, Scheffler A. Pentacyclic triterpene distribution in various plants—rich sources for a new group of multi-potent plant extracts. Molecules. 2009;14:2016–31.
Yogeeswari P, Sriram D. Betulinic acid and its derivatives: a review on their biological properties. Curr Med Chem. 2005;12(6):657–66.
Alakurtti S, Makela T, Koskimies S, Yli-Kauhaluoma J. Pharmacological properties of the ubiquitous natural product betulin. Eur J Pharm Sci. 2006;29(1):1–13.
Haque S, Nawrot DA, Alakurtti S, Ghemtio L, Yli-Kauhaluoma J, Tammela P. Screening and characterisation of antimicrobial properties of semisynthetic betulin derivatives. PLoS ONE. 2014;9(7):e102696. https://doi.org/10.1371/journal.pone.0102696.
Flekhter OB, Boreko EI, Nigmatullina LR, Tret’yakova EV, Pavlova NI, Baltina LA, Nikolaeva SN, Savinova OV, Eremin VF, Galin FZ, Tolstikov GA. Synthesis and antiviral activity of betulonic acid amides and conjugates with amino acids. Russ J Bioorg Chem. 2004;30:80–8.
Pavlova NI, Savinova OV, Nikolaeva SN, Boreko EI, Flekhter OB. Antiviral activity of betulin, betulinic and betulonic acids against some enveloped and non-enveloped viruses. Fitoterapia. 2003;74:489–92.
Flekhter OB, Ashavina OY, Boreko EI, Karachurina LT, Pavlova NI, Kabalnova NN, Savinova OV, Galin FZ, Nikolaeva SN, Zarudii FS, Baltina L, Tolstikov G. Synthesis of 3-O-acetylbetulinic and betulonic aldehydes according to Svern and the pharmacological activity of related oximes. Pharm Chem J. 2002;36:303–6.
Govdi AI, Sorokina IV, Tolstikova TG, Vasilevsky SF, Tolstikov GA. Synthesis and biological activity of novel acetylene betulonic acid derivatives. Chem Sustain Dev. 2010;18:397–402.
Vasilevsky SF, Govdi AI, Shults EE, Shakirov MM, Sorokina IV, Tolstikova TG, Baev DS, Tolstikov GA, Alabugin IV. Efficient synthesis of the first betulonic acid-acetylene hybrids and their hepatoprotective and anti-inflammatory activity. Bioorg Med Chem. 2009;17:5164–9.
Saxena BB, Zhu L, Hao M, Kisilis E, Katdare M, Oktem O, Bomshteyn A, Rathnam P. Boc-lysinated-betulonic acid: a potent, anti-prostate cancer agent. Bioorg Med Chem. 2006;14:6349–58.
Semenov DE, Zhukova NA, Ivanova EP, Sorokina IV, Baiev DS, Nepomnyashchikh GI, Tolstikova TG, Biryukova MS. Hepatoprotective properties of betulonic acid amide and heptral in toxic liver injury induced by carbon tetrachloride in combination with ethanol. Bull Exp Biol Med. 2015;158(3):336–41.
Dinh Ngoc T, Moons N, Kim Y, De Borggraeve W, Mashentseva A, Andrei G, Snoeck R, Balzarini J, Dehaen W. Synthesis of triterpenoid triazine derivatives from allobetulone and betulonic acid with biological activities. Bioorg Med Chem. 2014;22(13):3292–300.
Haque Shafiul, Nawrot Dorota A, Alakurtti Sami, Ghemtio Leo, Yli-Kauhaluoma Jari, Tammela Päivi. Screening and characterisation of antimicrobial properties of semisynthetic betulin derivatives. PLoS ONE. 2014;9(7):e102696. https://doi.org/10.1371/journal.pone.0102696.
Anikina LV, Tolmacheva IA, Vikharev YuB, Grishko VV. The immunotropic activity of lupane and oleanane 2,3-seco-triterpenoids. Russ J Bioorg Chem. 2010;36:240–4.
Sorokina IV, Tolstikova TG, Zhukova NA, Petrenko NI, Schults EE, Uzenkova NV, Grek OR, Pozdnyakova SV, Tolstikov GA. Betulonic acid and derivatives, a new group of agents reducing side effects of cytostatics. Dokl Biol Sci. 2004;339:434–7.
Loftsson T, Jarho P, Masson M, Jarvinen T. Cyclodextrins in drug delivery. Expert Opin Drug Deliv. 2005;2:335–51.
D’Aria F, Serri C, Niccoli M, Mayol L, Quagliariello V, Iaffaioli RV, Biondi M, Giancola C. Host–guest inclusion complex of quercetin and hydroxypropyl-β-cyclodextrin. J Therm Anal Calorim. 2017;130:451–6.
Garcia A, Leonardi D, Salazar MO, Lamas MC. Modified β-cyclodextrin inclusion complex to improve the physicochemical properties of albendazole. Complete in vitro evaluation and characterization. PLoS One. 2014;9(2):e88234. https://doi.org/10.1371/journal.pone.0088234.
Sbârcea L, Udrescu L, Ledeţi I, Szabadai Z, Fuliaş A, Sbârcea C. β-cyclodextrin inclusion complexes of lisinopril and zofenopril. Physicochemical characterization and compatibility study of lisnopril-β-cyclodextrin with lactose. J Therm Anal Calorim. 2016;123:2377–90.
Sbârcea L, Udrescu L, Drăgan L, Trandafirescu C, Szabadai Z, Bojiţă M. Fosinopril–cyclodextrin inclusion complexes: phase solubility and physicochemical analysis. Pharmazie. 2011;66:584–9.
Cȋrcioban D, Ledeţi A, Vlase G, Coricovac D, Moaca A, Farcaş C, Vlase T, Ledeţi I, Dehelean C. Guest–host interactions and complex formation for artemisinin with cyclodextrins: instrumental analysis and evaluation of biological activity. J Therm Anal Calorim. 2018;134:1375–84.
Brusnikina M, Silyukov O, Chislov M, Volkova T, Proshin A, Mazur A, Tolstoy P, Terekhova I. Effect of cyclodextrin complexation on solubility of novel anti-Alzheimer 1,2,4-thiadiazole derivative. J Therm Anal Calorim. 2017;130:443–50.
Sbârcea L, Udrescu L, Drăgan L, Trandafirescu C, Sasca V, Barvinschi P, Bojiţă M. Characterization of fosinopril natrium-hydroxypropil-β-cyclodextrin inclusion complex. Rev Chim Buchar. 2011;62(3):349–51.
Ikeda H, Fukushige Y, Matsubara T, Inenaga M, Kawahara M, Yukawa M, Fujisawa M, Yukawa E, Aki H. Improving water solubility of nateglinide by complexation of β-cyclodextrin. J Therm Anal Calorim. 2016;123:1847–50.
Sbârcea L, Ledeţi I, Drăgan L, Kurunczi L, Fuliaş A, Udrescu L. Fosinopril sodium–Hydroxypropyl-β-cyclodextrin inclusion complex. Thermal decomposition kinetics and compatibility studies. J Therm Anal Calorim. 2015;120:981–90.
Stella VJ, He Q. Cyclodextrins. Toxicol Pathol. 2008;36:30–42.
Laza-Knoerr AL, Gref R, Couvreur P. Cyclodextrin for drug delivery. J Drug Target. 2010;18:645–56.
Brusnikina M, Silyukov O, Chislov M, Volkova T, Proshin A, Terekhova I. New water-soluble dosage forms of 1,2,4-thiadiazole derivative on the basis of inclusion complexes with cyclodextrins. J Therm Anal Calorim. 2017;127:1815–24.
Fontanay S, Kedzierewicz F, Duval RE, Clarot I. Physicochemical and thermodynamic characterization of hydroxyl pentacyclic triterpenoid acid/γ-cyclodextrin inclusion complexes. J Incl Phenom Macrocycl Chem. 2012;73(1):341–7.
Şoica CM, Danciu C, Săvoiu-Balint G, Borcan F, Ambrus R, Zupko I, Bojin F, Coricovac D, Ciurlea S, Avram Ş, Dehelean CA, Olariu T, Matusz P. Betulinic acid in complex with a gamma-cyclodextrin derivative decreases proliferation and in vivo tumor development of non-metastatic and metastatic B164A5 Cells. Int J Mol Sci. 2014;15(5):8235–55.
Zong W, Bi SM. The preparation and characterization of inclusion complex of ursolic acid with γ-cyclodextrin. Adv Mater Res. 2012;403–408:712–6.
Ren Y, Liu Y, Yang Z, Niu R, Gao K, Yang B, Liao X, Zhang J. Solid inclusion complexes of oleanolic acid with amino-appended β-cyclodextrins (ACDs): preparation, characterization, water solubility and anticancer activity. Mater Sci Eng C. 2016;69:68–76.
Şoica CM, Peev CI, Ciurlea S, Ambrus R, Dehelean CA. Physico-chemical and toxicological evaluations of betulin and betulinic acid interactions with hydrophilic cyclodextrins. Farmacia. 2010;58(5):611–9.
Dehelean CA, Şoica CM, Peev CI, Gruia AT, Şeclaman E. Physico-chemical and molecular analysis of antitumoral pentacyclic triterpenes in complexation with gamma-cyclodextrin. Rev Chim Buchar. 2008;59:887–90.
Claude B, Morin Ph, Lafosse M, Andre P. Evaluation of apparent formation constants of pentacyclic triterpene acids complexes with derivatized β- and γ-cyclodextrins by reversed phase liquid chromatography. J Chromatogr A. 2004;1049:37–42.
Ledeti I, Bosca CS, Cosma C, Badea V, Todea A, Bercean VN. Study on obtaining 3-oxolup-20(29)-en-28-oic acid (betulonic acid) from (3β)-lup-20(29)-en-3,28-diol (betulin). Rev Chim Buchar. 2014;65(11):1289–93.
Chislov M, Silyukov O, Kumeev R, Proshin A, Perlovich G, Terekhova I. Complex formation of cyclodextrins with some pharmacologically active 1,2,4-thiadiazole derivatives. J Therm Anal Calorim. 2017;127:1797–805.
Benesi HA, Hildebrand JH. A spectrophotometric investigation of the interaction of iodine with aromatic hydrocarbons. J Am Chem Soc. 1949;71(8):2703–7.
Salústio J, Feio G, Figueirinhas JL, Pinto JF, Cabral Marques HM. The influence of the preparation methods on the inclusion of model drugs in a ß-cyclodextrin cavity. Eur J Pharm Biopharm. 2009;71:377–86.
Protein Data Bank. http://www.pdb.org/pdb/home/home.do. Accessed 20 Oct 2018
DeLano, W. L. PyMOL. DeLano Scientific, San Carlos, CA, 700, 2002.
Job P. Formation and stability of inorganic complexes in solution. Ann Chim. 1928;9:113–203.
Udrescu L, Sbârcea L, Fuliaş A, Ledeţi I, Vlase G, Barvinschi P, Kurunczi L. Physicochemical analysis and molecular modeling of the Fosinopril β-cyclodextrin inclusion complex. J Spectrosc (Spetrosc-Int J 2014) (2014):748468
Ledeţi I, Bercean V, Vlase G, Vlase T, Ledeţi A, Şuta L. Betulonic acid. Study of thermal degradation by kinetic approach. J Therm Anal Calorim. 2016;125:785–91.
Doile MM, Fortunato KA, Schműcker IC, Schucko SK, Silva MAS, Rodrigues PO. Physicochemical properties and dissolution studies of dexamethasone acetate- β-cyclodextrin inclusion complexes produced by different methods. AAPS Pharm Sci Tech. 2008;9:314–32.
Liu L, Zhu S. Preparation and characterization of inclusion complexes of prazosin hydrochloride with β-cyclodextrin and hydroxypropyl-β-cyclodextrin. J Pharm Biomed Anal. 2006;40:122–7.
Cîntă-Pînzaru S, Dehelean CA, Soica C, Culea M, Borcan F. Evaluation and differentiation of the Betulaceae birch bark species and their bioactive triterpene content using analytical FT-vibrational spectroscopy and GC-MS. Chem Cent J. 2012;6:67.
Lin-Vien D, Colthup NB, Fateley WG, Grasselli JG. The handbook of infrared and raman characteristic frequencies of organic molecules. New York: Academic Press; 1991.
Negi JS, Singh S. Spectroscopic investigation on the inclusion complex formation between amisulpride and –cyclodextrin. Carbohydr Polym. 2013;92:1835–43.
Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. Automated docking using a lamarckian genetic algorithm and empirical binding free energy function. J Comput Chem. 1998;19:1639–62.
Ledeţi I, Murariu M, Vlase G, Vlase T, Doca N, Ledeţi A, Şuta L-M, Olariu T. Investigation of thermal-induced decomposition of iodoform. J Therm Anal Calorim. 2017;127(1):565–70.
Suta LM, Vlase G, Ledeti A, Vlase T, Matusz P, Trandafirescu C, Circioban D, Olariu S, Ivan C, Murariu MS, Stelea L, Ledeti I. Solid-state thermal behaviour of cholic acid. Rev Chim. 2016;67(2):329–31.
Ledeti A, Olariu T, Caunii A, Vlase G, Circioban D, Baul B, Ledeti I, Vlase T, Murariu M. Evaluation of thermal stability and kinetic of degradation for levodopa in non-isothermal conditions. J Therm Anal Calorim. 2017;131(2):1881–8.
Suta LM, Matusz P, Ledeti A, Ivan C, Murariu M, Sora MC, Ledeti I. Embedding of biliary calculi in plastic materials a viable solution for increasing their mechanical resistance during sampling. Mater Plast. 2016;53(1):19–22.
Ledeti A, Vlase G, Vlase T, Bercean V, Murariu MS, Ledeti I, Suta LM. Solid-state preformulation studies of amiodarone hydrochloride. J Therm Anal Calorim. 2016;126(1):181–7.
Acknowledgements
This work was supported by a grant from the “Victor Babeş” University of Medicine and Pharmacy Timisoara (Grant PIII-C1-CFI-2014/2015-03 to L.S., A.L., L.U. and I.L.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sbârcea, L., Ledeţi, A., Udrescu, L. et al. Betulonic acid—cyclodextrins inclusion complexes. J Therm Anal Calorim 138, 2787–2797 (2019). https://doi.org/10.1007/s10973-019-08359-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-019-08359-6