
Abstract
A serial of tests were carried out to evaluate the effect of specimen mass on the test results for PMMA conducted in a micro-scale combustion calorimeter. Seven heating rates were used to test specimens of mass ranging from 0.5 to 6.0 mg with nominal interval of 0.5 mg. Eighty-five specimens were tested. Heat release rate, onset temperature, temperature at maximum heat release rate, total heat release, and heat release capacity were determined. The influence of specimen mass at each heating rate was analyzed. Specimen mass influences the maximum heat release rate, onset temperature, and temperature at maximum heat release rate significantly. The higher the heating rate, the greater the influence. Reliable results could be obtained as long as the specimen mass is more than 1 mg with oxygen concentration above 5 %; thus, the oxygen concentration limit might be extended from 10 to 5 %.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As noted in the Govmark® Micro-scale Combustion Calorimeter (MCC-2) data sheet [1], the Micro-scale Combustion Calorimeter was invented by the Federal Aviation Administration (FAA) to offer industry a research tool to assist the FAA in its mandate to dramatically improve the fire safety of aircraft materials. The MCC was developed as an efficient tool for screening new materials [2] and to provide a quantitative method to evaluate the effect of flame retardants in flame-retardant materials design [3]. Although the test results of the MCC do not include physical behavior such as melting, dripping, swelling, shrinking, delamination, and char/barrier formation that can influence the results of large (decagram/kilogram) samples in flame and fire tests [1], it can be used to develop models of flammability and prediction of fire behavior [4]. More and more material develop experts have used MCC as efficient tool to screen fire retardant [5–7].
This instrument has great potential in flammability prediction; however, its application may present several problems that need investigation. Specimen mass is one of the key parameters in performing a MCC test to provide a reliable result. MCC is becoming a mainstay in fire-testing laboratories due to its ability to obtain meaningful test data with a sample size in the range of 0.5–50 mg, in the so-called micro-scale term [1]. Govmark® [8] has specified that in “preparing a sample: 5 mg is the nominal recommended mass, but the normal mass for a sample is 3–10 mg.”
ASTM International has designated D7309 for this instrument as an ASTM standards publication [9–11]. The standards ASTM D7309-07 (2007) [8] and ASTM D7309-11 (2011) [10] have expired, while ASTM D7309-13 (2013) [11] is currently in effect. In ASTM D7309-13, section “9. Test Specimens” specifies “specimen mass shall be in the range of 1–10 mg. Specimen mass is subject to the constraint that oxidation of the specimen gases consumes less than one half of the available oxygen in the combustion gas stream at any time during the test and at the heating rate used in the test. Typical specimen mass is 2–5 mg.” The heating rates are normally in the range of 0.2–2 K s−1 depending on the specimen size. There has been an obvious change in the selection of sample mass and heating rate in Section 10.1.2.1 in ASTM D7309-13 compared to the earlier ASTM standards. In the expired ASTM D7309-07 (2007) [9] and ASTM D7309-11 (2011) [10], “to minimize temperature gradients within the sample the heating rate β for a sample of mass M 0 shall be β ≤ (5 mg K s−1)/M 0 in accordance with Ref. [12],” while in ASTM D7309-13 [1, 11], the choice of heating rate is associated with error allowance of tests [11, 13].
Lyon RE [13] established a thermokinetic model for the reaction rates of solids measured using constant heating rate differential thermal analysis derived from heat transfer and chemical kinetics and proposed an accuracy criterion for thermal analysis measurements that could be used for recommending experimental practice. In terms of the maximum reaction rate, the thermokinetic model provides a simple analytic relationship between the sample mass, heating rate, and measurement error for chemically reacting solids in non-isothermal analyses. This achievement provides a key criterion in ASTM D7309-13 for choosing specimen mass and heating rate. In Ref. [13], the relationship between specimen mass and heating rate with a measurement error ε due to thermal diffusion for thermal decomposition of typical polymers was given as,
where m 0 is initial specimen mass, β is heating rate, ε is measurement error, T p is the temperature at peak pyrolysis, Φ is a thermokinetic parameter defined as
where E a is activation energy, α is the thermal diffusivity of the solid in terms of its thermal conductivity κ and heat capacity c. R is general gas constant, and ρ is density of polymer.
Equation (1) could work as a criterion for the sample mass m 0 in terms of the maximum allowable measurement error ε at heating rate β in non-isothermal tests.
The present study focuses on the influence of specimen mass on experiment results and revisits the [13]’s error allowance using tests of PMMA (polymethylmethacrylate). PMMA is used as the reference material for fire calorimeters [14] because it thermally degrades to methylmethacrylate monomer in quantitative yield at the surface of the burning specimen, so the heat of combustion of the volatiles is a constant value. PMMA is considered as a representative of non-charring polymers [15] as there is no residence left.
In the present research, a correlation analysis between the specimen mass and the experimental results is carried out. The influences of specimen mass on maximum heat release rate, onset temperature, temperature at maximum heat release rate, and oxygen concentration limit were analyzed quantitatively. The research can provide guidance for the MCC experimental method in order to obtain more reliable data and also provide further understanding of flammability of PMMA.
Experimental methods
Facilities and material
MCC tests were conducted with a Govmark MCC-2 Microscale Combustion Calorimeter located at the VTT research center of Finland. Specifications of the Govmark MCC-2 instrument are as follows [1],
-
1.
Sample heating rate: 0–10 K s−1
-
2.
Gas flow rate: 50–200 cm3 min−1, response time of <0.1 s, sensitivity of 0.1 % of full scale,
-
3.
Repeatability is ±0.2 % of full scale and an accuracy of ±1 % of full scale deflection.
-
4.
Sample size: 0.5–50 mg (milligrams).
-
5.
Detection limit: 5 mW.
-
6.
Repeatability: ±2 % (10 mg specimen).
Pyrolyzer heating temperature was from 75 to 600 °C, and combustor temperature was set at 900 °C.
All tests followed the “Method A” procedure of the MCC. In the “Method A” procedure, the specimen undergoes a controlled thermal decomposition [11] when subjected to control heating in an oxygen-free/anaerobic environment. The gases released by the specimen during operation are swept from the specimen chamber by nitrogen, subsequently mixed with excess oxygen, and then completely oxidized in a high-temperature combustion furnace. The volumetric flow rate and volumetric oxygen concentration of the gas stream exiting the combustion furnace are continuously measured during the test to calculate the rate of heat release by means of oxygen consumption. In Method A, the heat of combustion of the volatile component of the specimen (specimen gases) is measured but not the heat of combustion of any solid residue [11]. As PMMA does not char, there is no residue left after test (Y p = 0), i.e., all solid volatilizes; thus, the measured heat of combustion of the volatile component of the specimen in our tests is the heat of combustion of PMMA, that is,
From the “Method A” procedure, the maximum heat release rate Q max, onset temperature T onset, temperature at maximum heat release rate T max, total heat release h c, heat release capacity η c, and oxygen concentration at maximum heat release rate \(\Delta_{{{\text{O}}_{ 2} }}\) may be determined. The parameter η c is defined as
where β is heating rate. It is considered that the flammability parameter η c is independent of the form, mass, and heating rate of the specimen as long as the specimen temperature is uniform at all times during the test [11].
The MCC tests were carried out for black PMMA ([C5H8O2]n) with density of 1180 kg m−3—a reference material for the cone calorimeter—and the MCC at VTT with thermal conductivity of 0.185 W m−1 K−1 and specific heat of 1.510 J g−1 K−1. There is no additive. It burns evenly and does not char in cone calorimeter test. Specimen mass was weighed by a Mettler AX205 AX-205 Analytical Semi Micro Balance Delta Range with readability of 0.01 mg in the weighing range of 81 g.
All tests were carried out according to the “repeatability conditions” [16] where independent test results are obtained with the same method on identical test items in the same laboratory by the same operator using the same equipment within short intervals of time.
Preparation of specimen and choice of heating rate
The specimen mass range requirement in ASTM D7309-13 is 1–10 mg with a recommended specimen mass range of 2–5 mg. From the technical sheet, the recommended specimen mass range of Govmark® is 0.5–50 mg. In the tests, seven heating rates, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, and 3.5 K s−1 were selected. Samples of different specimen mass from nominal mass of 0.5 mg with nominal interval 0.5 mg were tested under each heating rate, as listed in Table 2; 85 specimens were tested totally. For the high heating rate, the lowest oxygen concentration level was monitored.
Test results and analysis
Typical data from tests
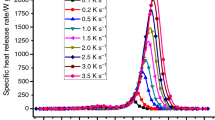
Figure 1 illustrates the heat release rate curves of 11 specimen masses, from 0.38–4.71 mg, at β = 3.5 K s−1.

Heat release rate curves at β = 3.5 K s−1
In some experiments (β = 3.5, 3.0, 2.5, 2.0, and 1.5 K s−1) with higher specimen mass, the oxygen concentration was lower than 10 %—the lowest being 1.29 %. In those cases, the corresponding Q max had a lower value than the average, while T max was higher than the average. The low Q max and high T max were caused by the extended process of pyrolysis due to an uneven heat transfer of larger specimen. The total heat release h c of these tests with low oxygen concentration was not reduced. This implies complete combustion occurs even for a low oxygen concentration. Typical data of the tests are shown in Table 1. The average Q max decreased with decreasing of heating rate, while T max increases with the increase in the heating rate, as occurs in a TG (thermogravimetric) tests on the same material [17].
At each heating rate, the Q max fluctuates within a certain range due to the different specimen masses, as shown in Table 1. The specimens with the highest Q max were mainly those with low mass (less than 1 mg) at each heating rate, except at β = 1.5 K s−1. However, the lowest Q max did not occur for the specimen with the most mass.
The heat release capacity η c changes from 304.72 ± 25.02 J g−1 K−1 (at β = 3.5 K s−1) to 577.49 ± 59.05 J g−1 K−1 (at β = 0.5 K s−1), while a relatively stable value of total heat release h c was obtained in the range 22.41–22.67 kJ g−1 at each heating rate.
The range of T max at each heating rate is listed in Table 2. The standard deviation of T max is less than 8 °C, and the average temperature decreases with increasing heating rate.
Correlation analysis
The correlation coefficient is a numerical measure of the strength of the relationship between two random variables [18]. The value of correlation coefficient varies from −1 to 1. A positive value means the two variables are positive correlated, that is the two variables vary in the same direction, while negative value indicates a negative correlation. A value close to +1 or −1 reveals the two variables are highly correlated. There are different types of correlation coefficient. Pearson’s product-moment correlation coefficient is the most widely used. It measures the linear relationship between two normally distributed variables. It was used by Lin TS [2] to study the correlations between MCC and conventional flammability tests for flame-retardant wire and cable compounds.
Pearson’s correlation coefficient is obtained by dividing the covariance of the two variables by the product of their standard deviations. The population correlation coefficient ρ X,Y between two random variables X and Y with expected values μ X and μ Y, and standard deviations σ X and σ Y is defined as:
where E is the expected value operator, cov means covariance, and corr a widely used alternative notation for Pearson’s correlation. The Pearson correlation is defined only if both of the standard deviations are finite, and both of them are nonzero.
For a series of n measurements of X and Y written as x i and y i where i = 1, 2,…, n, then the sample correlation coefficient can be used to estimate the population Pearson’s correlation r between X and Y. The sample correlation coefficient is written
where x and y are the sample means of X and Y, and s x and s y are the sample standard deviations of X and Y.
This can also be written as:
The Pearson correlation coefficient can vary from −1 (exact negative linear relation) to +1 (exact positive linear relation). The coefficient measures the strength of the linear relationship between two variables. For the following discussion, the strength of a correlation coefficient is arbitrarily defined in Table 3 [2].
Pearson’s correlation coefficients were calculated between specimen masses M 0 and onset temperature T onset, maximum HRR Q max, the temperature at maximum HRR T max, oxygen concentration \(\Delta_{{{\text{O}}_{ 2} }}\), and total heat release h c (Table 4). According to the definition of η c (Eq. 4), the Pearson correlation coefficients for mass M 0 and η c are the same as those for mass and Q max; thus, those were not included in Table 4.
M 0 has a negative linear relationship with Q max, the strength varies from weak (r xy = −0.44, β = 1.5 K s−1) to strong (r xy = −0.92, β = 3.0 K s−1). The range of correlation coefficients between M 0 and T max is from 0.54 to 0.80, and it does not show a strong relationship. M 0 has poor to marginal relationships with h c at all the seven heating rates, and the relationship tends to be weaker with the decrease in heating rate. M 0 has negative marginal (r xy = −0.61, β = 1.5 K s−1) to strong (r xy = −0.93, β = 2.0 K s−1) linear relationship with T onset. The results show a strong negative relationship with \(\Delta_{{{\text{O}}_{ 2} }}\) as the greater the mass the more oxygen consumed. The correlation analysis shows that specimen mass has a greater effect on Q max at higher heating rates.
General effect of specimen mass
The size of specimen has a significant influence on heat transfer and gas diffusion through the material and, therefore, some key parameters of the MCC are greatly affected as shown in Figs. 2–4. The rate of temperature rise for the interior of a specimen depends on the heating rate and the thermal capacity of the specimen. A greater mass of specimen results in more thermal capacity. It takes a shorter time for a smaller specimen to reach uniform temperature compared to that for a specimen with larger mass. Thus, a specimen with a smaller mass reaches the pyrolysis temperature overall faster than that with larger mass, and it results in a higher Q max and a lower T max. As shown in Fig. 2 the smaller the mass, the higher the Q max. The Pearson correlation coefficient between slope of fitting line and heating rate is −0.93. The slopes have strong negative linear relationship with heating rates.

Relationships between Q max and M 0
When a line-of-best-fit is performed on the Q max versus M 0 data it can be observed the slope of the line increases with an increase in heating rate. This implies the influence of mass on Q max is more significant at higher heating rates as shown in Fig. 2. In Fig. 3, the slopes of the lines-of-best-fit are similar which implies the effect of specimen mass on T max is similar at different heating rates. At higher heating rates, a higher T max is observed consistent with the results for thermogravimetry (TG) tests. Specimens with larger mass have a lower T onset and a higher T max as illustrated clearly in Figs. 3 and 4. A specimen with a larger mass has a larger surface area and produces more gaseous material at the initial heating stage resulting in a lower T onset. The Pearson correlation coefficient between slope of fitting line and heating rate in Fig. 3 is 0.58. The slopes have marginal linear relationship with heating rates. The Pearson correlation coefficient between slope of fitting line and heating rate in Fig. 4 is −0.82. The slopes have negative moderate linear relationship with heating rates.

Relationships between T max and M 0

Relationships between T onset and M 0
Relative error analysis of the tests
To illustrate the compound effect of heating rate β and specimen mass M 0 on the data, contour maps were drawn for the relative errors of Q max, T max, and h c.
Figure 5 is the contour map of the relative error of Q max with M 0 and β. The areas of high relative error are red and yellow in the contour map. As shown in Fig. 5, high relative error appears in the area of low and high specimen mass (M 0 < 1.5 mg and M 0 > 4.5 mg), and the corresponding β ranges are 0.5–1.25 and 1.75–3.5 K s−1. In the middle of contour is the blue region of low relative error (M 0 = 1.5–4.5 mg, β = 0.75–3.25 K s−1).

Contour map of relative error of Q max
Figure 6 is the contour map of relative error of T max with specimen mass and heating rate. Although the relative errors are different, the maximum value is less than 5 %. It implies that the relative error of T max is acceptable in present tests, and the choice of the heating rate and specimen mass has less effect on T max than on Q max.

Contour map of relative error of T max
Figure 7 is the contour map of relative error of h c with specimen mass and heating rate. Most of the area is blue which means the relative error is lower than 2.25 %. As PMMA volatilizes completely in the heating process, heating rate has little effect on h c. There are higher relative errors in the area of low specimen mass and low heating rate. From the signal point of view, there is a higher relative error in the low heating rate with low specimen mass areas due to a lower signal to noise ratio.

Contour map of relative error of h c
Oxygen concentration limitations
Figure 8 shows the oxygen concentration \(\Delta_{{{\text{O}}_{ 2} }}\) of the peak heat release rate with a line-of-best-fit. At the same heating rate, the greater the specimen mass, the more oxygen is consumed. Oxygen concentration \(\Delta_{{{\text{O}}_{ 2} }}\) is linear with specimen mass. The Pearson correlation coefficient between slope of fitting line and heating rate is −0.99. The slopes have strong negative linear relationship with heating rates. The relationship between oxygen concentration \(\Delta_{{{\text{O}}_{ 2} }}\) and specimen mass M 0 would be described by Eq. (8),
where slope(β) is the slope of a linear fit of data for \(\Delta_{{{\text{O}}_{ 2} }}\) versus M 0 at a certain heating rate. Then M 0 could be calculated by Eq. (9).

Oxygen concentration at Q max with different heating rate and sample mass
The slopes of the line-of-best-fit at different heating rates are shown in Fig. 9, and fit the linear equation, Eq. (10).

Linear fit of the slopes in Fig. 8
The slope of the relationship between \(\Delta_{{{\text{O}}_{ 2} }}\) and M 0 at a certain heating rate can be calculated by Eq. (10). Further substitution of that slope using Eq. (9) will determine the required specimen mass to ensure the oxygen concentration does not fall below a certain level at this heating rate. For example, the slope at heating rate β = 0.1 K s−1 is −0.633 % mg−1 calculated by Eq. (10). If the lowest oxygen concentration limit is set as 10 %, M 0 is 14.22 mg calculated by Eq. (9). For β = 1 K s−1, slope(β) is −1.38 % mg−1, then the M 0 is 6.52 mg (with the oxygen concentration limit 10 %), or 10.15 mg (with the oxygen concentration limit 5 %).
The activation energy E a of PMMA was determined by Kissinger plots [13] with T p at specific heating rate β, shown in Fig. 10. The thermokinetic parameter Φ is calculated for PMMA by Eq. (2) with α = 8.82 × 10−8 m2 s−1 [19], ρ = 1180 kg m−3=1.180 × 109 mg m−3, and E a = 164 kJ mol−1,

Kissinger plot of (β, T p) data to determine activation energy
This value is used in Eq. (1), i.e.,
The maximum specimen mass for a specified ε (0.05) at certain β is calculated by Eq. (9) for heating rates of 0.5,1.0, 1.5, 2.0, 2.5, 3.0 and 3.5 K s−1, and the corresponding average T p(β) listed in Table 1. The results are shown in Fig. 11 with the specimen mass limit obtained from oxygen concentration limitations.

The relationship of specimen mass and heating rate for certain oxygen concentrations
A specimen mass range is also shown in Fig. 11 calculated by Eq. (12) with typical polymer properties [13]: T p = 700 ± 70 K, α = 1.2 ± 0.2 × 10−7 m2 s−1, ρ = 1100 ± 150 kg m−3 = 1.1 ± 0.15 × 109 mg m−3, and E a = 200 ± 50 kJ mol−1,
The maximum specimen masses of PMMA at specified heating rates are calculated by Eq. (11) with ε = 0.05, and they are all within the range calculated by Eq. (12) for typical polymers, as shown in Fig. 11. Most of the maximum masses are less than 1 mg. But according to the analysis of relative error in Sect. 3.4, the specimens with mass less than 1 mg have high relative errors at all heating rates. The possible maximum specimen mass can also be calculated by using of oxygen concentration, and the range is 2.52 mg (3.5 K s−1) to 9.85 mg (0.5 K s−1) for 10 % minimum oxygen concentration and 5.88 mg (3.5 K s−1) to 19.63 mg (0.5 K s−1) for 1 % minimum oxygen concentration (refer Fig. 11). The latter range is obviously not desirable.
Conclusions
Based on tests conducted using PMMA and subsequent analysis, it was observed that specimen mass has significant influence on MCC results.
A suitable specimen mass should be determined for a MCC test in advance either by using a thermokinetic model [13] or by observation of the oxygen concentration and the shape of the heat release rate curve. When comparing the flammability of two materials with the MCC, besides using the same heating rate, the specimen mass must be chosen carefully. It is recommended to use the same amount of specimen masses to do comparison tests. A very small specimen mass (≤0.5 mg for PMMA) should be avoided. A very small specimen pyrolyzes especially fast and tends to exhibit an abnormal high Q max thus being prone to large error.
The correlation analysis shows that specimen mass has significant effect on the Q max at high-temperature rates. At lower heating rates, the influence of specimen mass on the peak heat release rate is less than that at a higher heating rate, which may be caused by different oxygen concentrations or heat transfer inside specimens. The lowest limit of oxygen concentration can be set at 5–10 %—within this range, there is no substantial effect on the results of h c and Q max.
When the same heating rate is used, there is a fluctuation in the specific heat release rate curve of specimens with larger mass. This fluctuation is not caused by incomplete combustion. This was because that the oxygen concentration of all tests was not depleted, and the total heat releases were all within a reasonable range. Uneven heat transfer in specimens with larger mass may be the main reason for the fluctuation.
Abbreviations
- α :
-
Thermal diffusivity (m−2 s−1)
- β :
-
Heating rate (K s−1)
- E a :
-
Activation energy (kJ mol−1)
- h c :
-
Net calorific value of sample (J g−1)
- h c,gas :
-
Specific heat of combustion of specimen gases (J g−1)
- η c :
-
Heat release capacity (J g−1 K−1)
- K :
-
Thermodynamic temperature (K)
- M o :
-
Initial specimen mass (mg)
- Mean:
-
Mean value of test data
- \(\Delta_{{{\text{O}}_{2} }}\) :
-
The change in the concentration (volume fraction) of O2 in the gas stream (%)
- ρ :
-
Density of oxygen at ambient conditions (kg m−3)
- Q(t):
-
Specific heat release rate at time t, (W g−1)
- Q max :
-
Maximum specific heat release rate, (W g−1)
- r xy :
-
Correlation coefficient
- R :
-
General gas constant (8.314 J mol−1 K−1)
- t :
-
Time (s)
- T :
-
Temperature (°C)
- T max :
-
Temperature of maximum peak heat release rate (°C)
- T onset :
-
Onset temperature of specific heat release rate (°C)
- T P :
-
Temperature of peak pyrolysis (K)
- T P (β):
-
Temperature of maximum pyrolysis at specified heating rate (K)
- Y p :
-
Pyrolysis residue (g g−1)
- σ :
-
Standard deviation
- Φ :
-
Thermokinetic parameter (K s mg−2/3)
References
Govmark Datasheet of Micro-scale Combustion Calorimeter (MCC2), the Govmark Organization, Inc.
Lin TS, Cogen JM, Lyon RE. Correlations between microscale combustion calorimetry and conventional flammability tests for flame retardant wire and cable compounds. International Wire and Cable Symposium, Proceedings of the 56th IWCS, November 11–14, 2007.
Yang CQ, He QL. Textile heat release properties measured by microscale combustion calorimetry: experimental repeatability. Fire Mater. 2012;36:127–37.
Matala A. Methods and applications of pyrolysis modeling for polymeric materials, Doctor degree dissertation, ISSN 2242-119x, VTT Science 44, Nov. 2013.
Xiao X, Hu S, Zhai JG, Chen T, Mai YY. Thermal properties and combustion behaviors of flame-retarded glass fiber-reinforced polyamide 6 with piperazine pyrophosphate and aluminum hypophosphite. J Therm Anal Calorim. 2016;125:175–85.
Majoni S. Thermal and flammability study of polystyrene composites containing magnesium–aluminum layered double hydroxide (MgAl-C16 LDH), and an organophosphate. J Therm Anal Calorim. 2015;120:1435–43.
Chen XY, Cai XF. Synthesis of poly(diethylenetriamine terephthalamide) and its application as a flame retardant for ABS. J Therm Anal Calorim. 2016;125:313–20.
Operation Manual, MCC Microscale Combustion Calorimeter, Govmark Organization, Inc. Version 1, Oct. 2010.
Standard Test Method for Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry, ASTM D7309-07, 2007.
Standard Test Method for Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry, ASTM D7309-11, 2011.
Standard Test Method for Determining Flammability Characteristics of Plastics and Other Solid Materials Using Microscale Combustion Calorimetry, ASTM D7309-13, 2013.
Lyon RE, Walters RN. Pyrolysis combustion flow calorimetry. J Anal Appl Pyrol. 2004;71:27–46.
Lyon RE, Safronava N, Senes J, Stoliarov SI. Thermokinetic model of sample response in nonisothermal analysis. Thermochimi Acta. 2012;545:82–9.
Lyon RE, Walters RN, Stoliarov SI, Safronava N. Principles and practice of microscale combustion calorimetry, DOT/FAA/TC-12/53, April 2013.
Stoliarov SI, Crowley S, Lyon RE, Linteris GT. Prediction of the burning rates of non-charring polymers. Combust Flame. 2009;156:1068–83.
Standard Practice for Use of the Terms Precision and Bias in ASTM Test Methods, ASTM E177-14, 2014.
Kannan P, Biernacki JJ, Visco DP Jr, Lambert W. Kinetics of thermal decomposition of expandable polystyrene in different gaseous environments. J Anal Appl Pyrol. 2009;84:139–44.
Correlation and dependence—Wikipedia, the free encyclopedia.mht. Accessed 20 Jan 2015.
Rhodes BT, Quintiere JG. Burning rate and flame heat flux for PMMA in a cone calorimeter. Fire Safety J. 1996;26:221–40.
Acknowledgements
This research is supported by the National Natural Science Foundation of China, No. 51376093. The research team is honored by “Six Talent Peaks” project of Jiangsu Province, China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Xu, Q., Jin, C., Griffin, G.J. et al. A PMMA flammability analysis using the MCC. J Therm Anal Calorim 126, 1831–1840 (2016). https://doi.org/10.1007/s10973-016-5688-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5688-z