
Abstract
The solid-state synthesis of undoped K0.5Na0.5NbO3 (KNN) and KNN doped with 1, 2 and 6 mol% Sr, from potassium, sodium and strontium carbonates with niobium pentoxide, was studied using thermal analysis and in situ high-temperature X-ray diffraction (HT-XRD). The thermogravimetry and the differential thermal analyses with evolved-gas analyses showed that the carbonates, which were previously reacted with the moisture in the air to form hydrogen carbonates, partly decomposed when heated to 200 °C. In the temperature interval where the reaction was observed, i.e., between 200 and 750 °C, all the samples exhibited the main mass loss in two steps. The first step starts at around 400 °C and finishes at 540 °C, and the second step has an onset at 540 °C and finishes with the end of the reaction between 630 and 675 °C, depending on the particle size distribution of the Nb2O5 precursor. According to the HT-XRD analysis, the perovskite phase is formed at 450 °C for all the samples, regardless of the Sr content. The formation of a polyniobate phase with a tetragonal tungsten bronze structure was detected by HT-XRD in the KNN with the largest amount of Sr dopant, i.e., 6 mol% of Sr, at 600 °C.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The piezoelectric effect is widely used in many applications, with the global market for piezoelectric devices being approximately $18.65 billion in 2013 [1]. The most commonly used material for these applications is lead zirconate titanate (PZT). Since 2003, when the European Union included PZT on its list of hazardous substances because of the toxic lead content [2, 3], the scientific community has been struggling to find a lead-free substitute [4]. In 2004, Saito et al. [5] reported on a lead-free piezoelectric ceramic based on a potassium sodium niobate solid solution with piezoelectric constants approaching those of PZT. Since then, the potassium sodium niobate solid-solution-based materials with a morphotropic phase boundary believed to be near the composition K0.5Na0.5NbO3 (KNN) became some of the most studied examples.
A high electromechanical coupling factor and a low dielectric permittivity make KNN-based ceramics interesting for ultrasonic applications [6]. However, KNN-based materials have a major drawback—the narrow sintering interval close to the melting point [7] makes KNN difficult to densify [8]. The most convenient way to improve the conventional solid-state sintering of KNN remains the doping of the ceramics. According to Jaffe [9], the densification should improve with A-site donor doping, i.e., keeping the charge within the unit cell neutral, which is to be compensated by creating vacancies on the A-site of the ABO3 perovskite. These vacancies would subsequently enhance the diffusion of ions during the sintering and improve the sinterability [9] because Sr2+ has an atomic radius (0.144 nm for CN = 12) right between the radii of K+ (0.164 nm for CN = 12) and Na+ (0.139 nm for CN = 12) [10] and therefore makes a suitable A-site dopant for KNN. Strontium doping improves both the density and the electrical properties of KNN when 0.5 and 1 % Sr are added [11–13]. In addition to the improved properties, there were other effects: With an increasing amount of Sr, there was a dramatic decrease in the grain size, a decrease in the peak splitting in the X-ray diffraction (XRD) pattern, a decrease in the phase-transition temperatures and the segregation of a secondary phase [12].
Another problem with KNN preparation is the reproducibility of the solid-state synthesis. Despite many reports on the preparation and properties of KNN, various authors who prepared KNN using the same procedure achieved quantitatively different functional properties [14–16]. This suggests that factors controlling the course of the solid-state synthesis are still not fully understood. In 2008, Malič et al. [17] reported on the solid-state reaction of a K2CO3 + Na2CO3/Nb2O5 diffusion couple at 600 °C. At first, the reaction layer made from a solid solution (KxNa1−x)2Nb4O11 is formed. The reaction proceeds by the diffusion of alkaline cations and oxygen anions via this reaction layer to form the K0.5Na0.5NbO3 perovskite phase on the interface of the intermediate phase and the mixture of carbonates. It was also concluded that the potassium ions, being the slower species, determine the growth of the reaction layer. Therefore, the particle size distribution of the starting powders will be crucial for the homogenization of the reagents and the size of the Nb2O5 will influence the time of the diffusion-controlled reaction [17]. Recently, we published a study that stresses again the importance of the solid-state synthesis precursors in KNN. Hreščak et al. [18] discovered that it is not only the particle size distribution of the Nb2O5 that is important, so is its polymorphic form. The as-received orthorhombic and monoclinic Nb2O5 were each used to prepare a batch of KNN in a solid-state synthesis with potassium and sodium carbonates. All of the precursors were wet planetary milled prior to the synthesis. Although the orthorhombic Nb2O5 remained one-phase after milling, the milling of the as-received monoclinic Nb2O5 resulted in a two-phase powder with orthorhombic nanocrystals attached to the surface of relatively larger monoclinic particles. While the as-received orthorhombic Nb2O5 reacted with the carbonates to form a homogenous KNN solid-state solution, the two-phased, as-received monoclinic Nb2O5 yielded an inhomogeneous KxNa1−xNbO3 solid solution with varying K/Na molar ratios. Based on these results, we decided to use orthorhombic Nb2O5 in this investigation.
It is often discussed that the potassium and sodium carbonates used for the solid-state synthesis are highly hygroscopic, which makes the solid-state synthesis of KNN even more challenging [19]. The rapid adsorption of water from the air is problematic when trying to achieve a stoichiometric composition [20]. Both drying at 200 °C and weighing in a dry atmosphere are necessary [19–21]. However, after the weighing, the homogenization and before the reaction with Nb2O5, the carbonates are again in contact with the humidity in the air and it is not clear how this influences the final phase composition.
The solid-state synthesis of different inorganic crystalline materials was previously successfully studied also by thermogravimetry combined with differential thermal and X-ray diffraction analyses [22, 23]. Therefore, to better understand the KNN processing, the solid-state synthesis of KNN and the influence of the Sr doping on the KNN’s reaction mechanism were studied using thermogravimetric analyses, differential thermal analyses and evolved-gas analyses, complemented with in situ high-temperature X-ray diffraction.
Experimental
Ceramic samples with the nominal formula (K0.5Na0.5)1−2ySryNbO3, for y = 0, 0.01, 0.02 and 0.06, subsequently denominated as KNN, KNN-1 % Sr, KNN-2 % Sr and KNN-6 % Sr, respectively, were prepared by solid-state synthesis from the following starting powders: K2CO3 (anhydrous, 99.9+ %, ChemPur, Karlsruhe, Germany), Na2CO3 (anhydrous, 99.9+ %, ChemPur, Karlsruhe, Germany), SrCO3 (99.994 %, Alfa Aesar, Karlsruhe, Germany) and Nb2O5 (325 mesh, 99.9 %, Sigma-Aldrich, Steinheim, Germany). All the carbonates and niobium pentoxides (except for the undoped KNN) precursors were dried for 6 h at 200 °C, ball-milled in a planetary mill in acetone for 4 h to achieve finer particles and a uniform particle size distribution and dried for 1 h at 105 °C and 2 h at 200 °C. The milled and dried precursors were weighed in the desired stoichiometric ratio in a dry box with an Ar atmosphere, and the mixtures were in a planetary mill for 4 h in acetone and dried for 1 h at 105 °C and 2 h at 200 °C. The reagents mixtures were then subjected to further analyses.
The thermogravimetry (TG), derivative thermogravimetry (DTG), differential thermal analysis (DTA) and evolved-gas analysis (EGA) were conducted on a Netzsch STA 409 C/CD coupled to a Balzers Quadstar 422 quadrupole mass spectrometer. Approximately 50 mg of the reagent mixture powder was placed in Pt/Rh crucibles and thermal changes were recorded in a dynamic flow of synthetic air at 100 mL min−1, with the following temperature program: First, the sample was heated to 200 °C with a 10 °C min−1 heating rate and held for 3 h in order to remove the water from the hygroscopic carbonate reagents. Then, the sample was heated to 750 °C with a heating rate of 10 °C min−1 to observe the reaction.
The phase-composition changes in the reagent mixtures upon heating were determined by high-temperature X-ray diffraction (HT-XRD) recorded at room temperature, 300, 400, 450, 500, 550, 600, 650, 700 °C, and once more at room temperature. The measurements were taken on a PANalytical X’Pert PRO MPD diffractometer with a Cu-Kα radiation source and a Ni-Kβ filter. The data were collected in the 2θ range from 20° to 40° with a step of 0.026°/100 s.
Granulometric analyses of the starting powders and reagent mixtures were performed with a Microtrac S3500 laser granulometer. The morphology of the reagents mixtures powders was analyzed using a Jeol JSM-7600F field-emission scanning electron microscope (SEM) equipped with energy-dispersive X-ray spectroscopy (EDXS).
Results
Figure 1 presents the TG-DTG-DTA-EGA of a representative sample, i.e., the reagents mixture corresponding to the KNN-1 % Sr formulation recorded between room temperature and 750 °C. Equation 1 summarizes the reaction of the carbonates with Nb2O5 to form the final Sr-doped potassium sodium niobate:

TG, DTG and DTA of the reagent mixture corresponding to KNN-1 % Sr complemented with an EGA analysis, in the temperature interval from room temperature to 750 °C
The temperature interval of the solid-state reaction in Fig. 1 is therefore between 200 and 750 °C, where the CO2 evolves in two steps (see Fig. 1 DTG and EGA) as a consequence of the carbonates’ decomposition [17, 20].
However, below 200 °C, the EGA shows that both water and CO2 are released from the system and are obviously accompanied by a certain mass loss, as seen in the TG curve. According to the DTA curve, this process is endothermic, with peaks at 121 and 194 °C.
Based on the Atlas of Thermoanalytical Curves [24], this process can be attributed to the decompositions of the sodium and potassium hydrogen carbonates, which take place below 200 °C, according to Eqs. 2a, 2b:
The first, smaller peak in the EGA curves could be attributed to the decomposition of NaHCO3, since according to the Atlas [24], NaHCO3 decomposes at 130 °C, while the second peak would then be the decomposition of KHCO3, peaking at 160 °C in the Atlas.
For the solid-state synthesis of KNN, alkali carbonates in an anhydrous form were used. After drying, they are weighed in a dry box, because they quickly adsorb water from the air. If weighed outside the dry box, the K2CO3 increases in mass much faster than the Na2CO3; therefore, the K2CO3 is expected to have much more water adsorbed on its surface. This is in agreement with the intensity of the EGA peaks and supports the attribution of the first peak as being the decomposition of NaHCO3 and the second as KHCO3.
Although the origin of the H2O in the hydrogen carbonates is obviously from the laboratory air, the origin of the simultaneous evolution of CO2 is not clear. To clarify whether the CO2 originated from the laboratory air or from the carbonate reagents, Table 1 compares the theoretical mass losses calculated based on Eq. 1 and the experimentally measured mass losses for all the samples, within different temperature intervals. The measured total mass loss in the interval 25–750 °C is higher than the theoretical value for all the samples. This is in agreement with the fact that the evolved water, originating from the air humidity, is also included in the total mass loss. When the temperature interval of the thermal decomposition of hydrogen carbonates is avoided, i.e., in the temperature interval 200–750 °C, the mass loss is lower than the theoretical value for all the samples.
The differences between the actual and the theoretical mass losses indicate that the CO2 evolving up to 200 °C also originates from the carbonates. The decomposition of the hydrogen carbonates, when heating or drying the powders, might influence the stoichiometry and therefore the phase purity of the final product. The phase composition of the powders is discussed later in Figs. 6 and 7.
In the subsequent text, the results of the samples’ behavior upon heating are only presented for the temperature interval 200–750 °C, in order to study the reaction of KNN doped with Sr.
Figure 2 presents the TG and DTG curves for KNN with 0, 1, 2 and 6 % Sr in the temperature interval between 200 and 750 °C. For all the samples, the main mass loss occurs in two steps revealed as two DTG peaks, with the first step between 400 and 540 °C and the second one between 540 °C and the end of the reaction. The latter varies from sample to sample, without any correlation with the Sr content (see Fig. 2, dotted lines). The reaction is finished at 675, 660, 630 and 660 °C for KNN, KNN-1 % Sr, KNN-2 % Sr and KNN-6 % Sr, respectively.

TG and DTG of reagents mixtures corresponding to the KNN with 0, 1, 2 and 6 % Sr in the temperature interval 200 to 750 °C. The full black lines mark the peak onset, the dotted lines mark the end of the reaction
Jenko [21] performed a thermal analysis of undoped-KNN reagent mixtures with different particle size distributions and observed that the temperature of the end of the reaction (subsequently denoted as T end) was increasing with the increasing particle size. In order to clarify the reason for the T end shift between the samples studied within this work, the particle size distributions of the reagents mixtures were investigated carefully, not only with a granulometric analysis, but also with SEM and EDXS analyses (Fig. 3). The median particle size (d 50) values from the granulometric analyses were 0.43, 0.53, 0.48 and 0.43 µm, for KNN, KNN-1, 2 and 6 % Sr, respectively, i.e., not correlating to the variations of T end.

BEI SEM images and granulometric analyses with the median particle size d 50 of the reagents mixtures prepared for KNN with 0, 1, 2 and 6 % Sr. The dark- and light-gray particles are Na2CO3 and K2CO3, respectively, while the white particles are Nb2O5. The SrCO3 has the light-gray contrast and could only be detected for KNN-6 % Sr
The backscattered electron images (BEIs) of the reagent mixtures were taken with an SEM, and the particles’ elemental compositions were qualitatively determined by EDXS analyses (see Fig. 3). No significant difference could be found among the samples’ particle morphologies. The largest, gray particles are the carbonates belonging to the particle size fraction 5–10 µm, as seen for the granulometric analyses. Carbonate particles in 1–5 µm fractions are also seen in the images. The dark-gray particles are the Na2CO3, and the light-gray particles are the K2CO3. The fine white particles belong to the Nb2O5. SrCO3 could be detected in the KNN-6 % Sr, as marked in Fig. 3—KNN-6 % Sr. Therefore, no correlation could be found regarding the particle size distribution or the particle morphology of the reagent mixtures and the end of the reaction.
The results of Malič et al. [17] also suggest that the time for the completion of the diffusion-controlled reaction between the alkali reagents and the niobium pentoxide would increase with the increasing particle size of the Nb2O5 reagent. Jenko [21] also conducted experiments with Nb2O5 using different particle size distributions, and the results support this suggestion. Jenko’s Nb2O5 had d 50 = 0.60 and 5.93 µm, the latter ending the reaction with carbonates at an almost 200 °C higher temperature.
Figure 4 and Table 2 present the granulometric analyses results of the Nb2O5 used for the synthesis of the studied samples. Table 2 also summarizes the T end from Fig. 2. Indeed, the highest T end (see Fig. 2—KNN), i.e., 675 °C, was measured for the KNN that was prepared from the Nb2O5 with the largest d 10, d 50, d 90 and a significantly larger portion of the particles above 1 µm in comparison with the other samples. The T end was the same, i.e., 660 °C for KNN-1 and 6 % Sr, which were prepared using the same batch of Nb2O5. Despite the Nb2O5 used for the preparation of KNN-1, 2 and 6 % Sr having comparable particle size distributions, there was a 30 °C difference in their T end.

Granulometric analyses of the Nb2O5 reagents used for the synthesis of KNN with 0, 1, 2 and 6 % Sr
The granulometric analyses mainly indicate that the T end shift cannot be correlated with the Sr content, but is characteristic for each batch of the KNN samples. The particle size distribution of the Nb2O5, however, remains the main factor causing the significant T end shifts.
Other mass-loss parameters that vary between the KNN samples include the intensity and the position of the two DTG peaks (see Fig. 2, DTG curves). For example, the ratios of the intensities of the two DTG peaks for KNN-2 % Sr and KNN-1 % Sr are opposite. The variation in the peaks’ intensities cannot be correlated with the Sr content. The unavoidable differences between the particle size distributions of Nb2O5 might also be the reason for the changing intensity of the DTG peaks, since the peaks’ intensity profiles are strongly related to the particular batch as well (see Fig. 2, intensity profile of the DTG peaks for KNN-1 % Sr and KNN-6 % Sr, prepared from the same batch of Nb2O5). Nevertheless, the typical calcination temperatures for KNN are 750–950 °C, which is safely above the end of the reaction for all the studied samples.
When carefully analyzing the DTA curves of all the samples in Fig. 5, one exothermic peak is observed in the same temperature interval as the first DTG peak, i.e., between 400 and 540 °C, and two DTA exothermic peaks are observed in the temperature interval of the second DGT peak, between 540 and ~675 °C, which is the temperature interval of the second DTG peak. The intensity of the corresponding exothermic peaks in the DTA curves varies depending on the varying intensity of the first and second DTG peaks. The first, second and third exothermic peaks appear at the same temperatures, i.e., at ~500, ~600 and ~650 °C, respectively, for KNN, KNN-1, and 2 % Sr. For the KNN-6 % Sr, the first peak is shifted to as low as 455 °C.

DTA of the reagent mixtures corresponding to KNN with 0, 1, 2 and 6 % Sr. Arrows mark the positions of the exothermic peaks
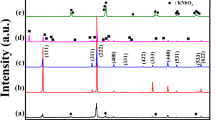
In order to be able to connect the thermally induced processes determined by the thermal analyses with the phase compositions of the powders, the HT-XRD was recorded as well. Figure 6 presents the HT-XRD spectra of all the studied reagent mixtures at different temperatures. The majority of the visible peaks of the unreacted powders at room temperature belong to the orthorhombic Nb2O5 (ICCD: 01-071-0336). Please note that the small peaks marked “*” at 25.7°, 35.3° and 37.9° 2θ belong to the Al2O3 sample holder (ICCD: 01-071-1128) and therefore are present with a variable intensity in all of the measured spectra. In the KNN-6 % Sr spectra the most intense SrCO3 peaks can also be identified (ICCD: 01-084-1778) (marked “s” in Fig. 6, KNN-6 % Sr). Above 400 °C, the intensity of the Nb2O5 peaks (marked “o” in Fig. 6) starts decreasing; at 450 °C the perovskite phase can be reliably detected, and its peaks (marked “P” in Fig. 6) increase with increasing temperature. As was previously reported, undoped KNN crystallizes in a monoclinic structure [16] and with the increasing Sr content, the unit-cell distortion decreases and the structure becomes pseudo-cubic above 2 % Sr [12]. The formation of the perovskite phase starts at the same temperature for all the samples, regardless of the Sr content. After heating the powders to 700 °C, the HT-XRD spectra of KNN, KNN-1 and 2 % Sr can be fully indexed according to the perovskite unit cell [16], which means that they are single phase. Besides the perovskite, a secondary phase was also detected in KNN-6 % Sr (see Fig. 6, KNN-6 % Sr, RT, marked with arrow). Figure 7 is the enlarged HT-XRD spectra of the KNN-6 % Sr to better show the appearance of the secondary phase peaks, which could be indexed with a polyniobate phase with a tetragonal tungsten bronze structure, e.g., K6Nb10.88O30 (ICCD 01-087-1856). The first appearance of the secondary-phase peaks was detected at 600 °C, but due to the low resolution of the HT-XRD spectra, it is possible that the secondary phase already appears at lower temperatures.

HT-XRD spectra of the reagent mixtures corresponding to KNN with 0, 1, 2 and 6 % Sr recorded at different temperatures. The peaks families are indexed with the KNN monoclinic unit cell according to Tellier [16]. Arrows mark the appearance of the perovskite phase (P) and the secondary phase peaks. Nb2O5 peaks are marked with o, and SrCO3 peaks are marked with s. The peaks marked with an asterisk belong to the Al2O3 sample holder

Enlarged HT-XRD of the reagent mixture corresponding to KNN-6 % Sr showing the peaks of the polyniobate secondary phase with the tetragonal tungsten bronze structure marked with arrows. The perovskite peak family {110} is indexed with the KNN monoclinic unit cell according to Tellier [16]. The Nb2O5 peaks are marked with o
In our study, the onset of the first DTA exothermic peak in KNN, KNN-1 and 2 % Sr is at 475 °C (Fig. 5), while the perovskite phase had already clearly crystallized at 450 °C (Fig. 6). The first, second and third exothermic peaks probably correspond to the combination of the processes happening during the KNN solid-state reaction, i.e., the formation of the intermediate phase (K x Na1−x )2Nb4O11 and because the potassium carbonate is expected to form the perovskite with niobium pentoxide at a different temperature than the sodium niobate [17]. The KNN samples doped with large amounts of Sr, i.e., above 2 % Sr content, tend to form a secondary potassium polyniobate phase, as mentioned by Malič et al. [12]. The secondary phase formation in the KNN-6 % Sr might be the reason for the sample’s modified DTA curve.
Conclusions
The solid-state syntheses of KNN and KNN doped with Sr were studied by thermal analyses complemented with evolved-gas analyses and in situ high-temperature X-ray diffraction (HT-XRD). The TG, DTA and EGA results from the temperature interval 25–200 °C, i.e., before the reaction starts, showed that despite the precautions the carbonates react with the moisture in the air and the decomposition of the formed hydrogen carbonates takes place.
During the reaction of the alkali (and strontium) carbonates with niobium oxide observed in the temperature interval 200–750 °C, all the samples exhibit a main mass loss that is characterized by the evolution of CO2 in two peaks with the same onset: the first at 400 °C and the second at 540 °C. The end of the reaction varies, but this was not correlated with the Sr content, rather with the differences in the Nb2O5 particle size distribution as the main factor influencing the temperature span of the reaction.
The DTA curves are similar for the KNN, KNN-1 and 2 % Sr, while the KNN-6 % Sr has a modified DTA curve, which could be related to the formation of the secondary phase in this sample.
The perovskite phase was detected by the HT-XRD at 450 °C in all the samples, regardless of the Sr content. The secondary-phase peaks could be detected by the HT-XRD in KNN-6 % Sr at 600 °C. Therefore, the only difference in the solid-state synthesis of KNN caused by the Sr doping was found to be that above 2 % Sr the secondary phase is formed, which then modifies the course of the reaction.
References
Acmite Market Intelligence. Global piezoelectric device market, market report. Ratingen: Acmite Market Intelligence; 2014.
EU-Directive 2002/96/EC. Waste electrical and electronic equipment (WEEE). Off J Eur Union. 2003;46(L37):24–38.
EU-Directive 2002/95/EC. Restriction of the use of certain hazardous substances in electrical and electronic equipment (RoHS). Off J Eur Union. 2003;46(L37):19–23.
Rödel J, Webber KG, Dittmer R, Jo W, Kimura M, Damjanovic D. Transferring lead-free piezoelectric ceramics into application. J Eur Ceram Soc. 2015;35(6):1659–81.
Saito Y, Takao H, Tani T, Nonoyama T, Takatori K, Homma T, et al. Lead-free piezoceramics. Nature. 2004;432(7013):84–7.
Safari A, Akdogan EK, editors. Piezoelectric and acoustic materials for transducer applications. New York: Springer; 2008.
Kosec M, Kolar D. On activated sintering and electrical properties of NaKNbO3. Mater Res Bull. 1975;10(5):335–9.
Malič B, Koruza J, Hreščak J, Bernard J, Wang K, Fisher JG, et al. Sintering of lead-free piezoelectric sodium potassium niobate ceramics. Materials. 2015;8:8117–46.
Jaffe B, Cook WR, Jaffe H. Piezoelectric ceramics. London: Academic Press; 1971.
Shannon RD. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr A. 1976;32(5):751–67.
Malic B, Bernard J, Holc J, Jenko D, Kosec M. Alkaline-earth doping in (K, Na)NbO3 based piezoceramics. J Eur Ceram Soc. 2005;25(12):2707–11.
Malic B, Bernard J, Holc J, Kosec M. Strontium doped K0.5Na0.5NbO3 based piezoceramics. Ferroelectrics. 2005;314:149–56.
Maeder MD, Damjanovic D, Setter N. Lead free piezoelectric materials. J Electroceram. 2004;13(1):385–92.
Acker J, Kungl H, Hoffmann MJ. Influence of alkaline and niobium excess on sintering and microstructure of sodium-potassium niobate (K0.5 Na0.5)NbO3. J Am Ceram Soc. 2010;93(5):1270–81.
Birol H, Damjanovic D, Setter N. Preparation and characterization of (K0.5Na0.5)NbO3 ceramics. J Eur Ceram Soc. 2006;26(6):861–6.
Tellier J, Malic B, Dkhil B, Jenko D, Cilensek J, Kosec M. Crystal structure and phase transitions of sodium potassium niobate perovskites. Solid State Sci. 2009;11(2):320–4.
Malic B, Jenko D, Holc J, Hrovat M, Kosec M. Synthesis of sodium potassium niobate: a diffusion couples study. J Am Ceram Soc. 2008;91(6):1916–22.
Hreščak J, Bencan A, Rojac T, Malič B. The influence of different niobium pentoxide precursors on the solid-state synthesis of potassium sodium niobate. J Eur Ceram Soc. 2013;33(15–16):3065–75.
Priya S, Nahm S, editors. Lead-free piezoelectrics. New York: Springer; 2012.
Malič B, Jenko D, Starowicz M, Bernard J, Kosec M, editors. Processing and characterization of (K, Na)NbO3 based piezoceramics. In: International conference on microelectronics, devices and materials, vol 38. Lipica, Slovenia: MIDEM—society for microelectronics, electronic components and materials; 2002.
Jenko D. Synthesis of (K, Na)NbO3 ceramics [Ph.D. thesis]. Ljubljana: University of Ljubljana; 2006.
Opravil T, Ptáček P, Šoukal F, Bartoníčková E, Wasserbauer J. Solid-state synthesis of SrY2O4 and SrSm2O4: mechanism and kinetics of synthesis, reactivity with water and thermal stability of products. J Therm Anal Calorim. 2016;123:181–94.
Arora C, Sharma A, Soni S, Naik Y, Ramarao G. Solid-state reaction of strontium oxalate with uranium oxalate: application of TG. J Therm Anal Calorim. 2016;124:43–9.
Liptay G. Atlas of thermoanalytical curves. Budapest: Akademiai Kiado; 1974.
Acknowledgements
This work was supported by the Slovenian Research Agency, under Grants P2-0105 and PR-03727. The authors would like to thank Edi Kranjc for measuring the HT-XRD of the samples.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Hreščak, J., Malič, B., Cilenšek, J. et al. Solid-state synthesis of undoped and Sr-doped K0.5Na0.5NbO3 . J Therm Anal Calorim 127, 129–136 (2017). https://doi.org/10.1007/s10973-016-5615-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5615-3