
Abstract
A series of copoly(ether-ester-urethane)s have been synthesized from poly(3-hydroxybutyrate) (PHB) diol prepolymer, as hard segments, and copoly(ε-caprolactone-ethylene glycol-ε-caprolactone) (PCL-PEG-PCL) diols with different PEG block lengths, as soft segments, with/without chitin whiskers (ChW) using hexamethylene diisocyanate, as a coupling agent, in one-step polymerization. The PHB content in the resulting copolymers was 0 and 40 %, and the content of ChW was varied from 0 to 5 %. The chemical structure of the resulting copolymers was characterized by FTIR, 1H-NMR and 13C-NMR spectra. The effect of chemical structure and ChW content on the thermal properties was studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TG). The DSC data revealed that the ΔH m of both PHB and PCL-PEG-PCL slightly increases with increasing the ChW content. The cold and melt crystallization of PHB enhanced with increasing ChW content. The TG data revealed that the thermal stability of copolymers slightly enhanced at high content of ChW. The swelling behavior of the copolymers was also investigated.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In recent years, biodegradable polymers have attracted attention due to their environmental compatibility and also diminishing the environmental waste disposal problems, which caused by the usage of traditional petroleum-derived plastics. Polyhydroxyalkanoates, of which poly(3-hydroxybutyrate) (PHB) is the most common member, are biodegradable polymers produced by many types of microorganisms from renewable sources such as sugar and molasses as intracellular storage material [1, 2]. Biocompatibility and biodegradability of PHB have attracted attention on tissue engineering and biomedical applications [3–7]. PHB is a highly crystalline polymer, has a high melting temperature and large spherulitic structures with slow poor mechanical properties [8]. Moreover, PHB is unstable at a temperature near to its melting temperature, which results in a drastic reduction in molecular weight during the melt processing [9, 10]. Many attempts have been done to modify PHB [11–21]. Block copolymerization can be an effective way to enhance PHB properties. Several researchers synthesized PHB block copolymers using telechelic hydroxyl-terminated PHB as hard segments, hydroxyl-terminated poly(ε-caprolactone) [22, 23], hydroxyl-terminated poly(butylene adipate) [24–26], or poly(ethylene glycol) (PEG) [27–29] as soft segments and diisocyanate as a coupling agent. They found that the physical properties and biodegradability can be adjusted by the proper choice of the used building hard and soft blocks, in addition to the hard/soft segments ratio. These copolymers possess much better physical properties such as lower melting points and higher extensibility compared to PHB homopolymer.
Poly(ε-caprolactone) (PCL) is one of the most frequently used building blocks for soft segments of degradable polyurethanes [30–35]. The labile aliphatic ester linkages of the PCL can be hydrolyzed and its degradation product, 6-hydroxyhexanoic acid, is a non-toxic metabolic product [32]. However, the rate of degradation of PCL is rather slow due to its hydrophobicity and semi-crystalline structure [36]. To obtain a better degradability and physical properties balance, the mixing of ester and ether polyol soft segments may be appropriate route for the preparation of copolymers [37]. PEG is introduced as comonomer for soft segment of polyurethane backbone. PEG presents many attractive properties, such as hydrophilicity, solubility in water and in organic solvents, nontoxicity and absence of antigenicity and immunogenicity, which allow it to be used for many clinical applications [37].
Recently, the introduction of reinforcing nanoparticles into a continuous polymer phase to form eco-friendly green nanocomposites has attracted a great deal of attention [38–44]. It can provide significant improvements in the mechanical properties, the thermal stability and can slow the biodegradation rate at very low volume fractions of the reinforcing phase [45–47]. The use of chitin nanowhisker (ChW) as a reinforcing nano-filler in the polymer matrix provides positive environmental benefits with respect to ultimate disposability, raw material use as well as strong mechanical properties.
The synthesis of biodegradable polyurethanes based on PCL and PEG diblock or triblock copolymers has been reported by many researchers [48–53]. They studied the relationship between the chemical structure of these polyurethanes and their biodegradation rates. They suggested that PCL usually enhances crystallinity and elastomeric mechanical properties of the polyurethanes, while PEG increases hydrophilicity and water absorption. Consequently, copolymerization of PHB-diol, as hard segments, and PCL-PEG-PCL triblock copolydiol, as soft segments, can offer the possibility to combine the advantages of these polymers in one copolymer. Our previous studies revealed that the synthesized polyurethanes based on PHB and PCL-PEG-PCL blocks exhibit better thermal and mechanical properties compared to PHB homopolymer [51]. ChW has excellent properties including biocompatibility, biodegradability, lack of toxicity, and advantageous absorption properties [54]. In this study our objective was to synthesize and characterize a group of biodegradable, amphiphilic poly(ether-ester-urethane)s and their ChW nanocomposites based on PHB, PCL-PEG-PCL triblock copolymer and 1,6-hexamethylene diisocyanate (HDI). The copolymer nanocomposites with different content of ChW were synthesized by in situ polymerization, and their thermal properties, morphology and water uptake were evaluated. It would be expected that the properties of the bionanocomposites can be improved; both in terms of mechanical properties and biocompatibility, over their pure polymer counter parts.
Experimental
Materials
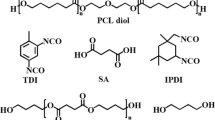
PHB (\( \bar{M}_{\text{n}} \) = 58,000 g mol−1, \( \bar{M}_{\text{w}} /\bar{\text{M}}_{\text n} = 2.3 \)) was supplied from Copersucar, Piracicaba (Brazil). For purification, PHB was dissolved in CHCl3, filtered to remove any insoluble matter, and precipitated in diethyl ether. PEG400 and PEG2000 (\( \bar{M}_{\text{n}} \) = 400 and 2000 g mol−1, respectively) were obtained from Aldrich and was dehydrated at 100–108 °C under vacuum for 48 h before use, ε-Caprolactone (ε-CL) was supplied from Aldrich and distilled under vacuum over calcium hydride and dried by 4 Å molecular sieves before use. Chitin was obtained from Funakashi Co. Ltd. (Japan). Dibutyltin dilaurate, 1,6-hexamethylene diisocyanate (HDI) and p-toluenesulfonic acid were supplied from Fluka. 1,3-Propanediol and N,N-dimethyl formamide (DMF) were purchased from Aldrich and purified before use.
Hydroxy-terminated PHB-diol (\( \bar{M}_{\text{n}} \) = 5000 g mol−1 with \( \bar{M}_{\text{w}} /\bar{M}_{\text{n}} \) = 2.4) was prepared by transesterification of high molecular weight PHB (\( \bar{M}_{\text{n}} \) = 58,000 g mol−1 and \( \bar{M}_{\text{w}} /\bar{M}_{\text{n}} \) = 2.3) with 1,3-propanediol in CHCl3 at 60 °C using p-toluenesulfonic acid as a catalyst according to the method described previously [23].
PCL-PEG400-PCL and PCL-PEG2000-PCL diols were prepared by ring opening polymerization of ε-CL initiated by PEG400 and PEG2000 using stannous octanoate as a reaction catalyst [37]. The synthesis is exemplified hereby for a triblock containing in average 10 ε-CL units. A typical PCL-PEG400-PCL-diol was synthesized as follows: 10 g (0.025 mol) of PEG400, 31.4 g (0.275 mol) of ε-CL and 0.21 g of Sn(Oct)2 (~0.5 mass% of total reactants) were charged into a reaction vessel under dry nitrogen atmosphere, and the reaction proceeded at 130 °C for 24 h, with stirring. The resulting copolymer was then cooled to room temperature, dissolved in dichloromethane, and reprecipitated from the filtrate using excess cold petroleum ether, filtered and dried under vacuum to constant mass at room temperature.
ChW suspension was prepared following the procedure described previously [55]. The purified chitin sample (1 g) was hydrolyzed in 3 mol L−1 HCl (30 mL) under reflux for 1.5 h with stirring. After acid hydrolysis, the suspension was diluted with distilled water followed by centrifugation (10,000 rpm) for 5 min. This process was repeated three times. Next, the suspension was dialyzed for several days against distilled water until a pH = 6 was achieved and then was freeze-dried. The mean diameter (d) of the prepared ChW is about 18 nm and the mean length (L) is 216 nm. Therefore, the aspect ratio (L/d) of ChW estimated from TEM (Fig. 1) is about 12.

TEM image of a dilute suspension of ChW
Synthesis of copoly(ether-ester-urethane)s and their ChW nanocomposites
A concentrated solution of the desired composition of PHB-diol and PCL-PEG-PCL-diol in freshly distilled DMF was introduced into a 100 mL round-bottom three-necked flask, equipped with a mechanical stirrer, a condenser and a nitrogen purge. A predetermined amount of HDI (NCO: OH = 1.05:1) and a catalytic amount of dibutyltin dilaurate were then added. The relative amounts of reaction mixture were adjusted so that the resulting copolymers would contain 0 and 40 % PHB. The reaction mixture was stirred at 120 °C under nitrogen atmosphere for 72 h. After polymerization was completed, the copolymers were separated in high yields (>95 %) by precipitation in excess amount of low boiling point petroleum ether. The isolated copolymers were purified by the dissolution in dioxane and reprecipitation in distilled water, filtered and dried under vacuum at room temperature for at least 48 h.
The copoly(ether-ester-urethane)s/ChW nanocomposites were prepared by adding the desired amount (2 and 5 %) of suspended ChW in 5 mL DMF to the same amount of HDI and the molar input ratios of PHB-diol/PCL-PEG-PCL-diol used in the preparation of neat samples; other experimental conditions were kept constant. The polymerization reaction is shown in Scheme 1, and the chemical compositions calculated from reactants feed ratios of the prepared samples are listed in Table 1.

Synthetic route of copoly(ether-ester-urethane)s
Water uptake
Water uptake of the copolymer samples and their nanocomposites was studied in distilled water at 30 °C. A known amount of pre-dried sample was placed into a flask with 25 mL water and kept undisturbed at 30 °C until equilibrium swelling was reached. After certain time (24 h), the swollen sample was taken off and its surface was quickly wiped off by absorbent paper just to remove the droplets on the surface, and then weighed.
where \( m_{\text{s}} \) and \( m_{\text{o}} \) are the masses of swollen and dried samples, respectively.
Characterization
FTIR spectra were recorded between 400 and 4000 cm−1 using a Perkin-Elmer B 25 spectrophotometer. All measurements were taken with 64 scans at resolution of 2 cm−1 at room temperature.
1H-NMR and 13C-NMR spectra were recorded on a Bruker AC-400 spectrophotometer in CDCl3. Non-deuterated CHCl3 was used as an internal reference.
SEM images were obtained using JEOL (JSM-5200) scanning electron microscope. Samples were prepared by placing small parts of films on a carbon tape on a stub and were coated with a thin layer of gold.
TEM image of ChW was obtained with Transmission Electron Microscope JEOL (JEM-1400 TEM) using an acceleration voltage of 100 kV. A drop of diluted suspension of ChW was deposited and dried on a carbon-coated grid before measuring.
Thermal degradation studies were conducted under nitrogen with dynamic heating rate of 10 °C min−1 using Shimadzu TGA-50 H Thermal Analyzer. All experiments were performed from room temperature to 600 °C, and the reference material was α-alumina. The sample masses in all experiments were taken in the range of 2–4 mg.
PL-DSC, (Polymer-Laboratories, England) Differential Scanning Calorimeter was employed to study the glass transition temperature (T g), melting temperature (T m), and crystallization behavior of the copolymers. The calorimeter was calibrated with ultra-pure indium. Samples (5–6 mg) were first heated from −30 to 160 °C with a heating rate of 10 °C min−1 (Run I). After keeping them at 160 °C for 2 min, samples were rapidly cooled to −60 °C at a rate of 80 °C min−1 to obtain specimen with low crystallinity, and then heated again with a heating rate of 10 °C min−1 to 160 °C (Run II). The melting temperature (T m) and the cold crystallization temperature (T cc) were taken as the peak values of the respective endotherm and exotherm processes in DSC curves. The apparent melting enthalpy (ΔH m) was determined from the area of the endothermic peaks. The glass transition temperature (T g) was taken as the midpoint of the specific heat capacity. The cooling curve run (III) was scanned over the temperature range from 160 to −60 °C at a constant rate of 10 °C min−1. The melt crystallization temperature (T mc) was determined from the exothermic peaks in this run. All measurements were taken under flow of nitrogen gas (25 mL min−1).
Results and discussion
Synthesis and characterization of the copolymers
Copoly(ether-ester-urethane)s were synthesized from PHB diol prepolymer (\( \bar{M}_{\text{n}} \) = 5000 g mol−1), as hard segments, and PCL-PEG-PCL diols with different PEG block lengths, as soft segments, with/without ChW using HDI as a coupling agent, in one-step polymerization as outlined in Scheme 1. The chemical structure of the synthesized copolymers was characterized by FTIR, 1H-NMR and 13C-NMR spectra. Typical FTIR spectra of the obtained copolymers and their corresponding prepolymers are shown in Fig. 2. All the characteristic absorptions bands of the PHB and the PCL-PEG-PCL segments were clearly recognized. The absorption band appearing at 1728 cm−1 is attributed to free carbonyl groups stretching of the PHB and PCL repeating units [56, 57]. –CH and ether stretching vibration bands assigned to the –OCH2CH2– repeating unit of PEG appear at 2883 and 1116 cm−1, respectively. Comparing the absorption bands of the prepared copolymers with their PHB and PCL-PEG-PCL prepolymers, it was noticed that the –OH absorption band appearing at 3500 cm−1, which is a characteristic band for diols, is disappeared, whereas two new characteristic absorption bands at 3400 and 1500 cm−1, belonging to –NH and –C–N stretching vibration modes of the urethane linkage, respectively, were observed. For copoly(ether-ester-urethane)s/ChW nanocomposites, the most important characteristics bands of PHB, PCL-PEG-PCL segments, urethane linkage and ChW can be seen in Fig. 2b. The increase in the intensity of the absorption band of the amide (II) of the nanocomposites relative to the carbonyl groups of PHB and PCL-PEG-PCL repeating units compared with that of pure sample is taken as evidence of the incorporation of ChW in copolymer matrix. The chemical structures of the prepared copolymers were confirmed by NMR spectroscopy. Typical 1H-NMR spectra of the copolymers A and B are shown in Fig. 3a, in which the signals of the protons of PHB, PCL-PEG-PCL segments and urethane linkage are assigned. The signals attributed to the PHB, repeating units (sample B) were observed at δ = 1.27 ppm due to the methyl, –O(CH)CH 3 CH2CO–, δ = 2.35–2.71 ppm due to methylene, –O(CH)CH3 CH 2 CO–, and δ = 5.21–5.30 ppm due to methine –O(CH)CH3CH2CO–, protons. The signal assigned at δ = 4.06 ppm is due to the methylene group next to the ester of the caprolactone units (–OCOCH 2 CH2CH2CH2CH2O–) and at δ = 3.60 is due to the methylene groups of the PEG repeating units (–CH 2 CH 2 O–). Figure 3b shows 13C-NMR spectra of copolymers A and B with their assignments peaks, the spectrum of A showed the characteristic signals of the carbons on the PCL at δ = 168, 73, 28, 23 and 20 ppm, PEG at δ = 63 and signals of carbons on the urethane linkage at δ = 168, 35, 24 and 22 ppm. For the copolymer B new signals appeared at δ = 173, 77, 40 and 19 ppm, which assigned to the carbons of PHB [20].

FTIR spectra: a prepolymer-diol, ChW, copolymers and their ChW nanocomposites; b B and B5%

a 1H-NMR spectra of the copolymers A and B. b 13C-NMR spectra of the copolymers A and B
Thermal properties of the copolymers
The thermal transition, crystallization, and melting behaviors of the copolymers with different compositions were initially studied by DSC. The values of glass transition temperature (T g), melting point (T m), fusion enthalpy (ΔH m) and cold crystallization temperature (T cc) were obtained from heating scans after rapid cooling (Run II), and melt crystallization temperature (T mc) was obtained from cooling scans (Run III). The results are summarized in Table 2.
Figure 4 shows the first DSC scans (Run I) of pure copolymer based only on PCL570-PEG400-PCL570 blocks (A) and that based on PHB and PCL570-PEG400-PCL570 blocks (B) and their nanocomposites with ChW. There is no significant change in the melting temperature of PCL-PEG-PCL and PHB segments with increasing the ChW content. Only one melting temperature is detected that corresponding to PHB, irrespective of PCL-PEG-PCL block length, for the neat copolymer B and its composite containing 2 mass% ChW (B2%), while two melting temperatures corresponding to PCL-PEG-PCL and PHB components appear for composite containing 4.7 mass% ChW (B5%). As seen in Table 2, the ΔH m of both PHB and PCL-PEG-PCL slightly increases with increasing the ChW content. The reheating DSC curves recorded after rapid cooling (Run II) of some copolymers and their ChW composites (Fig. 5), depict only one glass transition temperature, T g, ranges from −54.5 to −53.0 °C (Table 2) corresponding to PCL-PEG-PCL soft segments. As expected, the copolymers based on PHB and PCL-PEG-PCL (B and D) exhibit a higher T g compared with that based only on PCL-PEG-PCL (A). This is attributed to the increase in restriction imposed on the mobility of the soft segments by incorporation of rigid PHB component. On the other hand, the incorporation of ChW shows insignificant change in T g. Beyond T g, DSC curves show an exothermic crystallization peak in the range from −32.8 to −16.6 °C followed by two endothermic melting peaks, which corresponded to the melting of PHB and PCL-PEG-PCL segments. It is obvious that the cold crystallization temperature (T cc) shifts to lower temperature with increasing the ChW content, suggesting that the ChW acts as nucleating agent and enhances the crystallization rate from glassy state. No cold crystallization peak was detected for PHB segments. Therefore, the PHB hard segments in these materials, compared with neat PHB, are so easily crystallized that they even crystallize almost completely during rapid cooling. This behavior suggests that the flexibility of the PCL-PEG-PCL soft segments, and the incorporation of ChW may promote the crystallization of the PHB hard segments. Comparing the DSC curves recorded from free crystal melt (Fig. 6, Run III), it is obvious that neat copolymer based only on PCL570-PEG400-PCL570 blocks (A) and their composites with ChW exhibit melt crystallization peak at around −5 and −10 °C for neat sample A and composite containing 4.7 mass% ChW (A5%), respectively. This peak appears at around 4 °C for composite containing 2 % ChW (A2%). This suggests that the incorporation of low content of ChW enhances the rate of crystallization of the PCL-PEG-PCL component from molten state, while the copolymers containing relatively high content of ChW retards the crystallization rate. For copolymers based on PHB and PCL-PEG-PCL (B and D), one melt crystallization temperature, at relatively high temperature (around 98 °C), is observed. This is attributed to the melt crystallization of PHB hard segments. The melt crystallization of PCL-PEG-PCL segments is difficult to detect in this cooling condition. This may be ascribed to the restriction imposed on the crystallization of the PCL-PEG-PCL soft segments by PHB crystalline phase which is easy to crystalize upon cooling condition. It is interesting to observe that the incorporation of ChW in the copolymers has no significant change in T mc.

DSC scans (Run Ι) of copolymers A, B and their ChW nanocomposites

DSC scans (Run П) of the copolymers A, B, D and their ChW nanocomposites

DSC scans (Run Ш) of copolymers A, B, D and their ChW nanocomposites
Figure 7 illustrates the TG and DTG curves at a heating rate 10 °C min−1 for copolymers A and B and their ChW nanocomposites. From these curves, the thermal stability parameters including, the onset decomposition temperature, which is defined as the temperature at 5 mass% loss (T 5%), and the temperatures, at which maximum rate of mass loss occurs for the first and second degradation stages, designated as T 1max and T 2max, respectively, are given in Table 3. As can be seen, a copolymer A based on PCL-PEG-PCL blocks only possesses a single degradation stage, while copolymer B derived from PHB and PCL-PEG-PCL blocks, exhibits two main overlapped degradation steps. The first mass loss is assigned to the degradation of PHB segments as a consequence of its relatively low thermal stability, whereas the second mass loss is dominated by the thermal degradation of the PCL-PEG-PCL segments. Considering the temperature at which the thermal degradation starts (T 5%) as a criterion of the thermal stability of the prepared copolymers, it is obvious that the thermal stability of nanocomposites slightly enhanced at high content of ChW.

TG and DTG curves of copolymers A, B and their ChW nanocomposites
Morphology of the copolymers
Figure 8 shows the morphological features of fractured surfaces of the copolymer B and its nanocomposite containing 4.7 mass% ChW (B5%). The surface roughness increases with the incorporation of ChW in the copolymer matrix.

SEM images of fractured surfaces of copolymers: a B and b B5%
Water uptake of the copolymers
As shown in Table 3, the copolymer based on PCL-PEG-PCL blocks only (A) swells much more in the presence of ChW and the water uptake percentage increases with increasing the ChW percent. While the copolymer derived from PHB and PCL-PEG-PCL blocks, (B), swells much more than copolymer based on PCL-PEG-PCL blocks only (A), and the water uptake percentage decrease with increasing the ChW content.
Conclusions
Segmented biodegradable copoly(ether-ester-urethane)s were successfully synthesized through the chain extension of PHB-diol prepolymer, as hard segments, and PCL-PEG-PCL-diols with different PEG block lengths, as soft segments, with/without ChW using HDI, as a coupling agent, in one-step solution polymerization. The obtained copolymers were confirmed by FTIR, 1H-NMR and 13C-NMR. DSC analysis showed that the prepared copolymers are semicrystalline thermoplastics whose crystalline domains stem from PHB and PCL-PEG-PCL segments. The T m of PHB segments in the copolymers is decreased to ~130 °C, about 50 °C less than that of neat PHB; thus, the copolymers are considered to be better processable than neat PHB. The ChW, acts as nucleating agent, enhances the crystallization rate of PCL-PEG-PCL soft segments from glassy state. The incorporation of low content of ChW enhances the crystallization rate of the PCL-PEG-PCL component from molten state, while copolymers containing relatively high content of ChW retard the crystallization rate. The cold and melt crystallization of PHB enhanced with increasing ChW content. The TG analysis showed that the thermal stability of the copolymers slightly enhanced at high content of ChW.
In conclusion, the investigated copolymers seem to combine the criteria of biodegradability with improved thermal stability and wide processability window compared with high molecular weight PHB. In addition, such copolymers exhibit fast non isothermal melt crystallization to be excellent biodegradable polymers.
References
Yokoo T, Matsumoto K, Ooba T, Morimoto K, Taguchi S. Enhanced poly (3-hydroxybutyrate) production in transgenic tobacco BY-2 cells using engineered acetoacetyl-CoA reductase. Biosci Biotechnol Biochem. 2015;79:986–8.
Anderson AJ, Dawes EA. Occurrence, metabolism, metabolic role, and industrial uses of bacterial polyhydroxyalkanoates. Microbiol Rev. 1990;54:450–72.
Nair MB, Baranwal G, Vijayan P, Keyan KS, Jayakumar R. Composite hydrogel of chitosan–poly (hydroxybutyrate-co-valerate) with chondroitin sulfate nanoparticles for nucleus pulposus tissue engineering. Colloids Surf B Biointerfaces. 2015;136:84–92.
Parveez GKA, Bahariah B, Ayub NH, Masani MYA, Rasid OA, Tarmizi AH, Ishak Z. Production of polyhydroxybutyrate in oil palm (Elaeis guineensis Jacq.) mediated by microprojectile bombardment of PHB biosynthesis genes into embryogenic calli. Front Plant Sci. 2015;6:1–12.
Sznajder A, Pfeiffer D, Jendrossek D. Comparative proteome analysis reveals four novel polyhydroxybutyrate (PHB) granule-associated proteins in Ralstonia eutropha H16. Appl Environ Microbiol. 2015;8:1847–58.
Xu M, Qin J, Rao Z, You H, Zhang X, Yang T, Wang X, Xu Z. Effect of Polyhydroxybutyrate (PHB) storage on l-arginine production in recombinant Corynebacterium crenatum using coenzyme regulation. Microb Cell Fact. 2016;15:1–15.
Wu H, Wang H, Chen J, Chen G-Q. Effects of cascaded vgb promoters on poly (hydroxybutyrate)(PHB) synthesis by recombinant Escherichia coli grown micro-aerobically. Appl Microbiol Biotechnol. 2014;98:10013–21.
De Koning G, Lemstra P. Crystallization phenomena in bacterial poly [(R)-3-hydroxybutyrate]: 2. Embrittlement and rejuvenation. Polymer. 1993;34:4089–94.
Khanna S, Srivastava AK. Recent advances in microbial polyhydroxyalkanoates. Process Biochem. 2005;40:607–19.
Marchessault R, Okamura K, Su C. Physical properties of poly (β-hydroxy butyrate). II. Conformational aspects in solution. Macromolecules. 1970;3:735–40.
Wang L, Zhu W, Wang X, Chen X, Chen GQ, Xu K. Processability modifications of poly (3-hydroxybutyrate) by plasticizing, blending, and stabilizing. J Appl Polym Sci. 2008;107:166–73.
Hong S-G, Gau T-K, Huang S-C. Enhancement of the crystallization and thermal stability of polyhydroxybutyrate by polymeric additives. J Therm Anal Calorim. 2010;103:967–75.
Malinová L, Brožek J. Mixtures poly ((R)-3-hydroxybutyrate) and poly (l-lactic acid) subjected to DSC. J Therm Anal Calorim. 2010;103:653–60.
Wen X, Lu X, Peng Q, Zhu F, Zheng N. Crystallization behaviors and morphology of biodegradable poly (3-hydroxybutyrate-co-4-hydroxybutyrate). J Therm Anal Calorim. 2012;109:959–66.
Hong S-G, Hsu H-W, Ye M-T. Thermal properties and applications of low molecular weight polyhydroxybutyrate. J Therm Anal Calorim. 2013;111:1243–50.
Li H, Lu X, Yang H, Hu J. Non-isothermal crystallization of P (3HB-co-4HB)/PLA blends. J Therm Anal Calorim. 2015;122:817–29.
Zaldivar MP, Fernández NG, Peña CG, Paneque MR, Valentín SA. Synthesis and characterization of a new semi-interpenetrating polymer network hydrogel obtained by gamma radiations. J Therm Anal Calorim. 2011;106:725–30.
Kuntanoo K, Promkotra S, Kaewkannetra P (2015) Fabrication of novel polyhydroxybutyrate-co-hydroxyvalerate (PHBV) mixed with natural rubber latex. In: Key Engineering Materials: Trans Tech Publ, pp 404–408
Reis KC, Pereira L, Melo ICNA, Marconcini JM, Trugilho PF, Tonoli GHD. Particles of coffee wastes as reinforcement in polyhydroxybutyrate (PHB) based composites. Mater Res. 2015;18:546–52.
Wei L, McDonald AG, Stark NM. Grafting of bacterial polyhydroxybutyrate (PHB) onto cellulose via in situ reactive extrusion with dicumyl peroxide. Biomacromolecules. 2015;16:1040–9.
Silvino AC, da Silva JC. Preparation of blends of oligo ([R, S]-3-hydroxybutyrate) and poly ([R]-3-hydroxybutyrate): thermal properties and molecular dynamic studies. Polym Test. 2015;42:144–50.
Hirt TD, Neuenschwander P, Suter UW. Synthesis of degradable, biocompatible, and tough block-copolyesterurethanes. Macromol Chem Phys. 1996;197:4253–68.
Saad GR, Lee Y, Seliger H. Synthesis and characterization of biodegradable poly (ester-urethanes) based on bacterial poly (R-3-hydroxybutyrate). J Appl Polym Sci. 2002;83:703–18.
Saad GR. Calorimetric and dielectric study of the segmented biodegradable poly (ester-urethane) s based on bacterial poly [(R)-3-hydroxybutyrate]. Macromol Biosci. 2001;1:387–96.
Saad GR, Seliger H. Biodegradable copolymers based on bacterial poly ((R)-3-hydroxybutyrate): thermal and mechanical properties and biodegradation behaviour. Polym Degrad Stab. 2004;83:101–10.
Aziz MSA, Naguib HF, Saad GR. Non-isothermal crystallization kinetics of bacterial poly (3-hydroxybutyrate) in poly (3-hydroxybutyrate-co-butylene adipate) urethanes. Thermochim Acta. 2014;591:130–9.
Zhao Q, Cheng G, Li H, Ma X, Zhang L. Synthesis and characterization of biodegradable poly (3-hydroxybutyrate) and poly (ethylene glycol) multiblock copolymers. Polymer. 2005;46:10561–7.
Zhao Q, Cheng G. Preparation of biodegradable poly (3-hydroxybutyrate) and poly (ethylene glycol) multiblock copolymers. J Mater Sci. 2004;39:3829–31.
Loh XJ, Goh SH, Li J. Hydrolytic degradation and protein release studies of thermogelling polyurethane copolymers consisting of poly [(R)-3-hydroxybutyrate], poly (ethylene glycol), and poly (propylene glycol). Biomaterials. 2007;28:4113–23.
Zhang C, Zhang N, Wen X. Synthesis and characterization of biocompatible, degradable, light-curable, polyurethane-based elastic hydrogels. J Biomed Mater Res A. 2007;82:637–50.
Gorna K, Polowinski S, Gogolewski S. Synthesis and characterization of biodegradable poly (ϵ-caprolactone urethane) s. I. Effect of the polyol molecular weight, catalyst, and chain extender on the molecular and physical characteristics. J Polym Sci Pol Chem. 2002;40:156–70.
Marcos-Fernández A, Abraham GA, Valentín J, Román JS. Synthesis and characterization of biodegradable non-toxic poly (ester-urethane-urea) s based on poly (ε-caprolactone) and amino acid derivatives. Polymer. 2006;47:785–98.
Chan-Chan L, Solis-Correa R, Vargas-Coronado R, Cervantes-Uc J, Cauich-Rodríguez J, Quintana P, et al. Degradation studies on segmented polyurethanes prepared with HMDI, PCL and different chain extenders. Acta Biomater. 2010;6:2035–44.
Rueda-Larraz L, d’Arlas BF, Tercjak A, Ribes A, Mondragon I, Eceiza A. Synthesis and microstructure–mechanical property relationships of segmented polyurethanes based on a PCL–PTHF–PCL block copolymer as soft segment. Eur Polym J. 2009;45:2096–109.
Saad GR, Lee Y, Seliger H. Synthesis and thermal properties of biodegradable poly (ester-urethane) s based on chemo-synthetic poly [(R, S)-3-hydroxybutyrate]. Macromol Biosci. 2001;1:91–9.
Guan J, Fujimoto KL, Sacks MS, Wagner WR. Preparation and characterization of highly porous, biodegradable polyurethane scaffolds for soft tissue applications. Biomaterials. 2005;26:3961–71.
Piao L, Dai Z, Deng M, Chen X, Jing X. Synthesis and characterization of PCL/PEG/PCL triblock copolymers by using calcium catalyst. Polymer. 2003;44:2025–31.
Kaushik A, Singh M, Verma G. Green nanocomposites based on thermoplastic starch and steam exploded cellulose nanofibrils from wheat straw. Carbohyd Polym. 2010;82:337–45.
Naguib HF, Aziz MSA, Saad GR. Effect of organo-modified montmorillonite on thermal properties of bacterial poly (3-hydroxybutyrate). Polym Plast Technol. 2014;53:90–6.
Naguib H, Aziz MA, Saad G. Synthesis, morphology and thermal properties of polyurethanes nanocomposites based on poly (3-hydroxybutyrate) and organoclay. J Ind Chem. 2013;19:56–62.
Carli LN, Daitx TS, Guégan R, Giovanela M, Crespo JS, Mauler RS. Biopolymer nanocomposites based on poly (hydroxybutyrate-co-hydroxyvalerate) reinforced by a non-ionic organoclay. Polym Int. 2015;64:235–41.
Silverman T, Naffakh M, Marco C, Ellis G. Morphology and thermal properties of biodegradable poly (hydroxybutyrate-co-hydroxyvalerate)/tungsten disulphide inorganic nanotube nanocomposites. Mater Chem. 2016;170:145–53.
Zhijiang C, Yi X, Haizheng Y, Jia J, Liu Y. Poly (hydroxybutyrate)/cellulose acetate blend nanofiber scaffolds: preparation, characterization and cytocompatibility. Mater Sci Eng C. 2016;58:757–67.
Larsson M, Bergstrand A, Mesiah L, Van Vooren C, Larsson A. Nanocomposites of polyacrylic acid nanogels and biodegradable polyhydroxybutyrate for bone regeneration and drug delivery. J Nanomater. 2014;2014:1–9.
Cao X, Habibi Y, Magalhães WLE, Rojas OJ, Lucia LA. Cellulose nanocrystals-based nanocomposites: fruits of a novel biomass research and teaching platform. Curr Sci. 2011;100:1172–6.
Hong S-G, Huang S-C. Effect of modified silica on the crystallization and degradation of poly (3-hydroxybutyrate). J Therm Anal Calorim. 2015;119:1693–702.
Saad GR, Salama HE, Mohamed NA, Sabaa MW. Crystallization and thermal properties of biodegradable polyurethanes based on poly [(R)-3-hydroxybutyrate] and their composites with chitin whiskers. J Appl Polym Sci. 2014;131:40784–96.
Jiang X, Li J, Ding M, Tan H, Ling Q, Zhong Y, Fu Q. Synthesis and degradation of nontoxic biodegradable waterborne polyurethanes elastomer with poly (ε-caprolactone) and poly (ethylene glycol) as soft segment. Eur Polym J. 2007;43:1838–46.
Yeganeh H, Jamshidi H, Jamshidi S. Synthesis and properties of novel biodegradable poly (ε-caprolactone)/poly (ethylene glycol)-based polyurethane elastomers. Polym Int. 2007;56:41–9.
Xie Z, Lu C, Chen X, Chen L, Hu X, Shi Q, et al. A facile approach to biodegradable poly (ε-caprolactone)-poly (ethylene glycol)-based polyurethanes containing pendant amino groups. Eur Polym J. 2007;43:2080–7.
Naguib HF, Aziz MSA, Sherif SM, Saad GR. Synthesis and thermal characterization of poly (ester-ether urethane) s based on PHB and PCL-PEG-PCL blocks. J Polym Res. 2011;18:1217–27.
Loh XJ, Sng KBC, Li J. Synthesis and water-swelling of thermo-responsive poly (ester urethane) s containing poly (ε-caprolactone), poly (ethylene glycol) and poly (propylene glycol). Biomaterials. 2008;29:3185–94.
Endres T, Zheng M, Kılıç A, Turowska A, Beck-Broichsitter M, Renz H, Merkel OM, Kissel T. Amphiphilic biodegradable PEG-PCL-PEI triblock copolymers for FRET-capable in vitro and in vivo delivery of siRNA and quantum dots. Mol Pharm. 2014;11:1273–81.
Gopalan Nair K, Dufresne A. Crab shell chitin whisker reinforced natural rubber nanocomposites. 1. Processing and swelling behavior. Biomacromolecules. 2003;4:657–65.
Bordes P, Pollet E, Avérous L. Nano-biocomposites: biodegradable polyester/nanoclay systems. Prog Polym Sci. 2009;34:125–55.
Coleman MM, Skrovanek DJ, Hu J, Painter PC. Hydrogen bonding in polymer blends. 1. FTIR studies of urethane-ether blends. Macromolecules. 1988;21:59–65.
Srichatrapimuk VW, Cooper SL. Infrared thermal analysis of polyurethane block polymers. J Macromol Sci B. 1978;15:267–311.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mohamed, N.A., Salama, H.E., Sabaa, M.W. et al. Synthesis and characterization of biodegradable copoly(ether-ester-urethane)s and their chitin whisker nanocomposites. J Therm Anal Calorim 125, 163–173 (2016). https://doi.org/10.1007/s10973-016-5388-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5388-8