
Abstract
Compatibility studies comprise an important step in pre-formulation since they allow the identification of the excipients most compatible with herbal extracts from different analytical techniques. The objective of this work is to evaluate the compatibility between the nebulized extract of S. brasiliensis Engler with pharmaceutical excipients using analytical techniques associated with chemometric tools. The extract was nebulized through aspersion and produced from the hydroalcoholic extract of the bark of S. brasiliensis Engler. Binary mixtures were produced in various proportions using the following pharmaceutical excipients: starch, microcrystalline cellulose (Avicel® 101 and 102), lactose, magnesium stearate, PVP K-30 and talc. The samples were analyzed by optical microscopy, differential scanning calorimetry and X-ray diffraction (XRD). With the data obtained from DSC curves, matrices for hierarchical cluster analysis (HCA) and principal component analysis (PCA) were made. Using microscopy, an amorphous formation and/or crystalline components could be seen. In DSC curves, as well as in PCA and HCA analyses, possible interactions were identified with starch, lactose and magnesium stearate. This was confirmed by XRD. The starch showed the greatest interaction. The results indicate that the DSC technique associated with chemometric tools contributed to a better interpretation of compatibility studies and that microcrystalline cellulose, PVP K-30 and talc were the most compatible excipients in relation to the extract.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
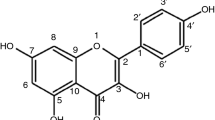
Schinopsis brasiliensis Engler is a plant belonging to the Anacardiaceae family. It is found in the Brazilian semiarid region, where it is known as “braúna” or “baraúna.” Studies show the antimicrobial activity of its extracts against strains of Pseudomonas aeruginosa, Staphylococcus aureus, Escherichia coli, Klebsiella pneumonia and Candida albicans [1–3]. Phytochemical analysis of the bark of S. brasiliensis allowed for the isolation of a new alkylphenol and indicated the presence of phenolic compounds, such as gallic acid and methyl gallate [4, 5].
The extract of S. brasiliensis was characterized by Fernandes [6] using thermal analysis, infrared spectroscopy, X-ray diffraction and scanning electron microscopy (SEM). The results suggested that the extract is stable, with the potential production of a phytotherapic medicine.
Phytotherapic medicines were obtained exclusively from plant-originated raw material. They are recognized either by clinical studies or by ethnopharmacological and technical–scientific knowledge. These products must meet the requirements of the market while maintaining efficacy, safety and quality. Thus, pre-formulation studies, especially compatibility tests, are the starting point for the development of stable medicines with therapeutic efficacy in terms of organoleptic properties, polymorphism, crystallinity/amorphicity and release time, among others [7, 8].
Different techniques are used to evaluate potential drug–excipient interactions, including dry extracts of medicinal plants used as drugs, among which optical microscopy, X-ray diffraction (XRD) and thermal analysis stand out [9–12].
Optical microscopy, although it is a simple technique, allows the visualization of the morphology of active ingredients. Detoisien [13] used, among other techniques, optical microscopy in polymorphic crystal visualizations. Alves-Silva [12] used optical microscopy to visualize itraconazole/benzinidazole compatibility with different excipients.
X-ray diffraction is a complementary technique to study drug–excipient compatibility. Its applicability consist in the fact that crystalline solids have a defined set of curve peaks while amorphous solids show a set of unordered curve peaks. An interaction can be observed through a modification or a suppression of curve peaks or changes in amorphous/crystal conditions [14]. Aiming to obtain the best dry extract by aspersion of Rhamnus purshiana, Gallo [15] used X-ray diffraction to characterize the product that proved to be a completely amorphous extract.
In terms of thermal analysis, differential scanning calorimetry (DSC) and thermogravimetry (TG) are the most common methods for analyzing the extracts and active substances of medicinal plants. Medeiros [16] evaluated the presence of colloidal silicon dioxide and cyclodextrin while drying Albizia inopinata using TG and photovisual DSC. The extract with cyclodextrin showed to be more stable in both techniques. Costa [17], using DSC and TG, assessed the compatibility of a lyophilized extract of Heliotropium indicum with hydroxyethyl cellulose, methyl paraben and propylene glycol; these last two substances interacted with the extract.
The thermoanalytical techniques are used in the extracts and excipient compatibility study, but due to the complex composition of the mixture, the observation of possible interactions between samples is difficult [17]. Thus, the application of chemometric techniques allows for a better interpretation of the results, enabling a further interpretation of the information from compatibility studies, which cannot be confirmed with a simple analysis of isolated results [18].
Different tools, such as principal component analysis (PCA) and hierarchical cluster analysis (HCA), are simple chemometric techniques that are used in several studies related to analytical techniques. Wesolowski [19], applied PCA on thermogravimetric analysis data and elemental chemical analytical, obtained from leaves and flowers and classified 44 different plant species. Melo [20] applied PCA on near-infrared spectroscopy (NIR) for the classification of pill samples containing dipyrone, caffeine and orphenadrine citrate.
In this context, the aim of this work is to evaluate the compatibility between the nebulized extract of S. brasiliensis with pharmaceutical excipients used in solid forms through analytical techniques using chemometric tools of pattern recognition.
Materials and methods
Herbal material and extract production
The stem bark of S. brasiliensis was collected from the region of Campina Grande, state of Paraiba (7 13′ 50′′ S, 35 52′ 52′′ W). The voucher specimen was prepared and identified by Professor Jayme Coelho de Morais Herbarium (PRU), Federal University of Paraiba under the EAN-14049.
The herbal material was dried at 40 °C using an air oven. After drying, the material was reduced in a knife mill (10-mesh outlet sieve) to a fine powder. The hydroalcoholic extract was obtained by maceration of the powder in a water:ethanol (30:70 v v−1) solvent system for 72 h and then dried by Spray Dryer (LabPlant®), with onset temperature at 140 °C and endset between 90 and 95 °C, under 3.0 mL min−1 flow rate. Colloidal silicon dioxide (Aerosil®200) was used as a drying agent at 20 % (w w−1). The drying extract was weighted, protected from light and stored in closed flasks at room temperature.
Binary mixtures
Binary mixtures were prepared from physical mixtures of the nebulized extract (BRCA) with the following pharmaceutical excipients: starch (CS), microcrystalline cellulose 101 (MC 101) and 102 (MC 102), lactose (LA), talc (TALC), magnesium stearate (MS) and polyvinylpyrrolidone K-30 (PVP K-30). The studies were performed using mixtures in proportions of 1:1, 1:2, 1:4, 1:6, 1:8 and 1:10 for starch, microcrystalline cellulose and lactose, and 1:1, 1:2 and 2:1 for talc, magnesium stearate and PVP K-30 based on their percentage of use in solid formulations [21]. The pharmaceutical excipients tested were all purchased from Brazilian industrial chemicals and pharmaceuticals products.
Optical microscopy
Optical microscopy was performed without prior treatment of the mixture in the proportion of 1:1 using a video microscope Hirox®, model KH-7700, with a 1400× increase.
Differential scanning calorimetry (DSC)
The DSC curves were obtained on a TA Instruments Calorimeter, model DSC Q20, using aluminum crucibles with about 2 ± 0.1 mg of samples under nitrogen atmosphere, at the flow of 50 mL min−1. Rising-temperature experiments were conducted at the temperature range from 25 to 400 °C and heating rate of 10 °C min−1. Indium (m.p. 156.6 °C) was used as standard for equipment calibration. Data were analyzed using the software TA Instruments Universal Analysis 2000, 4.7A.
Chemometric analysis
The calorimetric data, excipient and binary mixture, were subjected to the following chemometric pattern recognition techniques: hierarchical cluster analysis (HCA), using Statistica 6.0 software; and principal component analysis (PCA), using The Unscrambler® 9.8 software. For a better view of the relations among the extract and excipients, the data were preprocessed by standard of normal variation (SNV), which is a transformation that removes scattering effects and that centers and scales each curve individually.
X-ray diffraction
The analyses of X-ray diffraction were carried out in a Shimadzu® diffractometer, model XRD-600. The scanning angle (2θ) was from 2° to 70°, with a rate of 2° min−1 and a Cu system (Kα1). The equipment operated in a 40.00 kV voltage and with a 30 mA current. Data were analyzed using Origin® software version 8.0.
Results and discussion
Optical microscopy
Figure 1 shows digital images of the extract BRCA and its mixtures with the excipients (1:1). Figure 1a displays the particles with a dark brown uneven staining of dry extract of BRCA. In Fig. 1b, it is possible to identify the starch grains, and in Fig. 1c, d, cellulose crystals were observed. Figure 1e, f shows the presence of crystalline regions, typical of lactose and magnesium stearate excipients, respectively. In PVP K-30 (Fig. 1g), some spherical particles were noticeable in the mixture, possibly from the povidone production process. However, regarding talc (Fig. 1h), it is possible to see its crystalline aspect [21].

Optical microscopy of the extract BRCA a and its binary mixtures: b starch, c microcrystalline cellulose 101, d microcrystalline cellulose 102, e lactose, f magnesium stearate, g PVP K-30 and h talc
Differential scanning calorimetry (DSC)
Figure 2 shows the DSC curve of the extract and of binary mixtures, and Table 1 shows the values of thermal events that were observed in DSC curves. The BRCA extract curve (Fig. 2a) showed three endotherm peaks. The most significant was at 69.06 °C (ΔH = 89.96 J g−1), possibly related to the loss of water, solvent (ethanol) or volatile compounds. The curve ends with the beginning of the sample degradation process, which was completed above 400 °C. This behavior was completely different from that found by Fernandes [6] with S. brasiliensis curves, but without the presence of drying aids, which may suggest that there is a greater stability of the dry extract with the addition of the drying agent.

DSC curves of the extract BRCA a and its binary mixtures: b starch, c microcrystalline cellulose 101, d microcrystalline cellulose 102, e lactose, f magnesium stearate, g PVP K-30 and h talc
In the mixture with starch (Fig. 2b), a possible interaction between the extract and the excipient occurred, and with the increase of starch proportion in the mixture, there was a change in heat peaks. Considering the mixtures 1:1 and 1:2, there was a suppression of peaks related to starch; only the extract characteristics were visible. A process between 73 and 76 °C was also observed, which is probably related to the gelatinization of cornstarch and its degradation above 300 °C. In the following mixtures, there were peaks related to starch, however, these peaks were shown at intensities completely different from pure starch [21].
It was observed that in microcrystalline cellulose (101 and 102), no changes were present in DSC curves (Fig. 2c, d) when compared to extract, mixtures and excipients. There were only small variations in heat flux dehydration. Types of celluloses differ based on aspects related to their physical characteristics, such as particle size.
Figure 2e shows the DSC curves of lactose. The first was related to dehydration at 147.64 °C, and the second was an endothermic event, possibly from the melting process anticipation of α-lactose at 217.47 °C, ΔH = 117.90 J g−1, followed by its decomposition. In mixture 1:1, there was the suppression of the melting peak of lactose and the sample degraded without fusion. In mixtures 1:2 and 1:4, a small endothermic event happened that culminated in the degradation of the sample. In mixtures 1:6, 1:8 and 1:10, an endothermic peak occurred with temperatures differing from the original lactose peak at 224.79, 227.51 and 229.15 °C, respectively. Macedo [22] and Medeiros [9], studying the compatibility between propranolol and prednisone with lactose, noted a possible interaction of lactose with the drugs.
The typical magnesium stearate curve (Fig. 2f) showed loss of water around 100 °C, an endothermic peak with the possibly of melting at 120.63 °C, and degradation at 349.57 °C. The literature reported that the magnesium stearate melting range is 117–150 °C. In the three mixtures, there is a suppression of the melting range and an anticipation of sample degradation. The interaction of the magnesium stearate was described by Bazzo [23] in studies with captopril and by Tita [24] in studies with acetylsalicylic acid.
In the mixtures with PVP K-30 (Fig. 2g) and talc (Fig. 2h), no significant changes were observed in the curves if compared when the excipients isolated.
Chemometric analysis
In a thermal characterization of the drug–excipient interaction, the mixture curve will be the result of individual curves of the components involved. The suppression, creation or movement of thermal events in relation to the drug or the excipient is considered to be a possible interaction [14]. Thus, the use of chemometric techniques to rapidly and easily reduce information in graphs and in interaction evaluation tables is necessary due to the large amount of data and the need for comparison of mixture graphs with different proportions. Thus, with the data from DSC curves, matrices were constructed and analyzed using pattern recognition chemometric models, such as HCA (hierarchical cluster analysis) and PCA (principal component analysis).
Figure 3 shows the HCA of the extract, excipients and mixtures of BRCA. In the graph of starch (Fig. 3a), the formation of two major groups were observed; the first extract contained the mixtures 1:1 and 1:2 at a 45 % bond distance, and the other with starch and other mixtures at 86 % bond distance. Despite this, it was possible to observe that the extract contributes little in DSC analyses using high starch proportions. It is worth mentioning that there is a greater similarity between the proportions of 1:1 and 1:2 for BRCA over other mixtures, which more closely resemble the excipient. Therefore, it is possible to infer an extract–excipient interaction by the disposition of mixtures closer to the excipient in dendograms, in particular 1:4, 1:6, 1:8 and 1:10.

Hierarchical cluster analysis (HCA) dendrograms of binary mixtures: a starch, b microcrystalline cellulose 101, c microcrystalline cellulose 102, d lactose, e magnesium stearate, f PVP K-30 and g talc
In the mixture with celluloses, there was a difference between 101 (Fig. 3b) and 102 (Fig. 3c). While with the cellulose 101 the formation of two major groups occurs, in cellulose 102 the mixtures resemble the excipient very strongly, isolating the BRCA. The microcrystalline cellulose 101 that is, the first group, formed at a 64 % bond distance contained 1:1, 1:2 and BRCA mixtures, similar to starch. The other group is formed by the other mixtures and by the pure excipient similar to the effect caused by starch with a 92 % bond distance proportion. The same consideration made regarding starch in the occurrence of the extract–excipient interaction applied to cellulose 101. In microcrystalline cellulose 102 (Fig. 3c), the mixtures with the pure excipient bind at a bond distance of 36 % for 1:4, 1:6, 1:8 and 1:10 form a group that binds with the group of mixtures 1:1 and 1:2 with a 50 % bond proportion. Thus, similar to previous dendograms, no organization of disposition of extract–excipient proportions occurred.
For lactose (Fig. 3d), the mixtures were distributed in accordance with the excipient. The percent bond distance between the mixtures 1:1, 1:2 and 1:4 for the excipient isolated occurred at 63 %, whereas in 1:6, 1:8 and 1:10 it happened at 74 %.
Analyzing the figures relating to the PVP K-30 (Fig. 3e) and magnesium stearate (Fig. 3f), it is possible to visualize the difference of organization of dendograms, when there are proportional mixtures (1:1, 1:2 and 2:1). In the case of PVP, the mixtures 1:1 and 1:2 are closer to the BRCA at a 29 % bond distance proportion, and the mixture 2:1 was at 52 % from the group that contains the excipient. On the other hand, considering magnesium stearate, two large groups appear, again isolating the excipient. Wesolowski and Rojek [18] found a similar organizational dendrogram. Upon examination, using HCA, the interaction occurred between atenolol and beta-cyclodextrin. The bond distance between BRCA and the mixture 2:1 to groups 1:1 and 1:2 was 62 %.
With the talc (Fig. 3g), there was the formation of two groups, one BRCA and the other with and the mixtures and the excipient. Initially, the mixtures 1:1 and 1:2 are samples closer in terms of distance, then the excipient and finally the mixture 2:1. This suggests that BRCA is closer to the mixture 2:1 than to the others, which seems to be an influence of the extract–excipient interaction.
In PCA with starch (Fig. 4a), the formation of one BRCA group containing the mixtures 1:1 and 1:2, another group with the mixtures 1:4 and 1:10, and a third group with starch and the mixture 1:8 and isolated 1:6 suggests an interaction by the spatial arrangement of groups [25]. It is possible to see the absence of anomalous samples in the leverage graph (Fig. 5a); DSC curves of BRCA, starch and the mixture 1:8 are the highest contributors in the spatial arrangement of the samples. The mixtures 1:6 and 1:10 are those with higher residues. This distribution of mixtures in the two-dimensional space PC1 × PC2, with 47 and 19 % of explained variance, respectively, points to an extract–excipient interaction in mixtures with a higher proportion of excipient, corroborating the HCA.

Principal component analysis (PCA) scores of binary mixtures: a starch, b microcrystalline cellulose 101, c microcrystalline cellulose 102, d lactose, e magnesium stearate, f PVP K-30 and g talc

Leverage graphs for the binary mixtures: a starch, b microcrystalline cellulose 101, c microcrystalline cellulose 102, d lactose, e magnesium stearate, f PVP K-30 and g talc
With celluloses 101 (Fig. 4b) and 102 (Fig. 4c), a group is formed among mixtures with the extract and the excipient remaining. The PC1 × PC2 explained variance for each of the excipients was 54 and 35 % for cellulose 101 and 80 and 12 % for cellulose 102. Moreover, when observing distances in both cases, a proximity between the extract and mixture 1:1 and between the excipient and the mixture 1:10 was noted. Wesolowski and Rojek [18] found a similar organization of a similar PCA in the mixture with atenolol and chitosan where no interaction was observed. This differs from the information presented in corresponding dendrograms to each of the excipients. This difference occurs because the HCA technique allows for the visualization of the degree of similarity among samples only by calculating distances using all the multidimensional space, whereas the PCA technique allows a design of new axes with respect to the most relevant information for each one of the variables in this multidimensional space. The same situation occurred with cellulose 102, with a greater distance from the extract in relation to mixtures and proximity to the excipient.
The score graph for PC1 × PC2 lactose (Fig. 4d), with 38 and 30 % of explained variance, respectively, resulted in one excipient group with the mixtures 1:1, 1:2 and 1:4, another with the mixtures 1:6, 1:8 and 1:10, and the isolation of BRCA, suggesting that the mixtures were not similar to the extract. In the mixture with magnesium stearate (Fig. 4e), only the mixture 2:1 had a certain proximity with the BRCA. It was found that both cases suggest interactions.
With PVP K-30 (Fig. 4f), in accordance with the PC1 × PC2 score graph, with 86 and 10 % of explained variance, respectively, the mixtures 1:1 and 1:2 were close to excipient, and the mixture 2:1 was isolated. In the score graph, talc (Fig. 4g) and PC1 (85 %) × PC2 (12 %) showed a mixture group and the separation of extracts, similar to what occurred with the HCA. Although they show such grouping, thermal data (Table 1) do not point to an interaction, and the group must have occurred due to a high similarity between the mixtures.
In terms of the leverage graph, it is possible to see that a similar situation occurred with the magnesium stearate, with a uniform distribution of mixtures, excipient and extract, in terms of their influences and the residue from the PCA model. It was observed that only the data that were significant in the model were analyzed. Regarding cellulose 101 (Fig. 5b), it was noted that the two mixtures that have a higher residue after the model was designed were 1:1 and 1:2. The other mixtures, excipients and the extract have a high influence on the model, excluding the mixture 1:6, which has a low influence and also a low residue. For cellulose 102 (Fig. 5c), the leverage graph shows a high influence of the mixtures 1:1, 1:2 and 1:8 in addition to the excipient and the extract and low influence and a high residue for other mixtures. This suggests, as shown with the HCA, an extract–excipient interaction with low proportions related to the excipient.
In the leverage graph (Fig. 5d) to lactose, the mixtures 1:1, 1:2 and 1:4 had a high residue and a low influence in model designing and other mixtures, but the excipient and the extract have a high influence in the model. For the magnesium stearate, the leverage graph (Fig. 5e) showed a homogeneous distribution of the importance of all components. Thus, the excipients that showed a greater interaction (starch, lactose and magnesium stearate) were subjected to X-ray diffraction (XRD) in 1:1 binary mixtures.
Figure 6a shows the XRD pattern of the BRCA extract, in which the amorphicity of the sample, common in aspersion-dried plant extracts, was visualized. A greater inclination in initial diffraction angles forming an amorphous aureole is also perceptible [26]. This is possibly due to the presence of sugars, which prevented the extract from returning to its crystalline form during the drying process instead organizing in an amorphous form. This phenomenon is common during the drying of fruit pulps using aspersion [27]. Fernandes [6] evaluated the X-ray diffraction of the dried extract of S. brasiliensis without the presence of a drying adjuvant. In both extracts, the conformation was amorphous, formed by a high noise, suggesting that the presence of the adjuvant does not affect the amorphous appearance of the extract by spray drying. Gallo [28] found a similar conformation with Hamamelis virginiana extract, which was dried with colloidal silicon dioxide and sodium carboxymethylcellulose.

X-Ray Diffraction curves of the extract and its binary mixtures: a BRCA, b starch, c lactose, d magnesium stearate
In Fig. 6b–d, it was observed that the binary mixtures were produced with starch, lactose and magnesium stearate excipients, respectively. In all of them, intensity changes were observed in curve peaks, which may suggest an interaction [24]. However, in the mixture with starch (Fig. 6d), besides the intensity change, a change in the orientation of initial and final angles was observed, although the final appearance remained that of an amorphous mixture, suggesting an incompatibility with the excipient [14].
Conclusions
Optical microscopy, although it is a simple technique, allows for the characterization of amorphous and crystalline solids. However, it is a supplementary technique in the compatibility study. On the other hand, the DSC technique associated with chemometric tools is very helpful in identifying incompatibility, especially in more complex active ingredients such as dry medicinal herbal extracts. Based on these data, interactions of BRCA extract with starch, lactose and magnesium stearate were identified due to changes in thermal events and changes in HCA and PCA graphs. The incompatibilities have been confirmed by X-ray diffraction, according to which starch had higher interactions.
In this context, microcrystalline celluloses 101 and 102, talc and PVP K-30 are the most suitable excipients for the further development of solid formulations with S. brasiliensis Engler.
References
Chaves TP, Dantas IC, Felismino DC, Vieira KVM, Clementino ELC, Costa LS. Atividade antimicrobiana das folhas de Schinopsis brasiliensis Engl. Biofar. 2011;5:11–7.
Saraiva AM, Castro RHA, Cordeiro RP, Peixoto-Sobrinho TJS, Castro VTNA, Amorim ELC, Xavier HS, Pisciottano MNC. In vitro evaluation of antioxidant, antimicrobial and toxicity properties of extracts of Schinopsis brasiliensis Engl. (Anacardiaceae). Afr J Pharm Pharmacol. 2011;5:1724–31.
Silva MSP, Brandão DO, Chaves TP, Formiga Filho ALN, Costa EMMB, Santos VL, Medeiros ACD. Study bioprospecting of medicinal plant extracts of the semiarid northeast: contribution to the control of oral microorganisms. Evid-Based Compl Alt. 2012;. doi:10.1155/2012/681207.
Cardoso MP, David JM, David JP. A new alkylphenol from Schinopsis brasiliensis Engl. Nat Prod Res. 2005;19:431–3.
Saraiva AM, Saraiva CL, Cordeiro RP, Soares RR, Xavier HR, Caetano N. Atividade antimicrobiana e sinérgica das frações das folhas de Schinopsis brasiliensis Engl. frente a clones multirresistentes de Staphylococcus aureus. Rev Bras Plantas Med. 2013;15:199–207.
Fernandes FHA, Santana CP, Santos RL, Correia LP, Conceição MM, Macêdo RO, Medeiros ACD. Thermal characterization of dried extract of medicinal plant by DSC and analytical techniques. J Therm Anal Calorim. 2013;113:443–7.
Gupta MM, Saini TR. Preformulation parameters characterization to design, development and formulation of vancomycin hydrochloride tablets for pseudomembranous colitis. Int J Pharm Res Dev. 2002;1:1–7.
Narang AS, Desai D, Badawy S. Impact of excipient interactions on solid dosage form stability. Pharm Res. 2012;29:2660–83.
Medeiros ACD, Cervantes NAB, Gomes APB, Macêdo RO. Thermal stability of prednisone drug and tablets. J Therm Anal Calorim. 2001;64:745–50.
Mura P, Furlanetto S, Cirri M, Maestrelli M, Marras AM, Pinzautti S. Optimization of glibenclamide tablet composition through the combined use of differential scanning calorimetry and d-optimal mixture experimental design. J Pharm Biomed Anal. 2005;37:65–71.
Tita B, Fulias A, Bandur G, Marian E, Tita D. Compatibility study between ketoprofen and pharmaceutical excipients used in solid dosage forms. J Pharm Biomed Anal. 2011;56:221–7.
Alves-Silva L, Sá-Barreto LCL. LIMA EM, Cunha-Filho MSS. Preformulation studies of itraconazole associated with benznidazole and pharmaceutical excipients. Thermochim Acta. 2014;575:29–33.
Detoisien T, Arnoux M, Taulelle P, Colson D, Klein JP, Veesler S. Thermal analysis: a further step in characterizing solid forms obtained by screening crystallization of an API. Int J Pharm. 2012;403:29–36.
Chadha R, Bhandari S. Drug-excipient compatibility screening—Role of thermoanalytical and spectroscopic techniques. J Pharm Biomed Anal. 2014;87:82–97.
Gallo L, Llabot JM, Allemandi D, Bucalá V, Piña J. Influence of spray-drying operating conditions on Rhamnus purshiana (Cáscara sagrada) extract powder physical properties. Powder Technol. 2011;208:205–14.
Medeiros ACD, Medeiros IA, Macêdo RO. Thermal studies of Albizia inopinata crude extract in the presence of cyclodextrin and Aerosil® by TG and DSC coupled to the photovisual system. Thermochim Acta. 2002;392–393:93–8.
Costa RS, Negrão CAB, Camelo SRP, Ribeiro-Costa RM, Barbosa WLR, Costa CEF, Silva Júnior JOS. Investigation of thermal behavior of Heliotropium indicum L. lyophilized extract by TG and DSC. J Therm Anal Calorim. 2015;111:1959–64.
Wesolowski M, Rojek B. Thermogravimetric detection of incompatibilities between atenolol and excipients using multivariate techniques. J Therm Anal Calorim. 2013;113:169–77.
Wesolowski M, Rojek B. Thermal decomposition and elemental composition of medicinal plant materials–leaves and flowers principal component analysis of the results. Thermochim Acta. 2003;397:171–80.
Melo CAD, Silva P, Gomes AA, Fernandes DD, Veras G, Medeiros ACD. Classification of tablets containing dipyrone, caffeine and orphenadrine by near infrared spectroscopy and chemometric tools. J Braz Chem Soc. 2015;24:991–7.
Rowe RC, Sheskey PJ, Quinn ME. Handbook of pharmaceutical excipients. 6th ed. Chicago: Apha; 2009.
Macedo RO, Do Nascimento TP, Veras JW. Compatibility and stability studies of propranolol hydrochloride binary mixtures and tablets for TG and DSC-photovisual. J Therm Anal Calorim. 2002;67:483–9.
Bazzo GC, Silva MAS. Estudo termoanalítico de comprimidos revestidos contendo captopril através de termogravimetria (TG) e calorimetria exploratória diferencial (DSC). Rev Bras Ciênc Farmac. 2005;41:319–22.
Tita B, Jurca T, Fulias A, Marian E, Tita B. Compatibility study of the acetylsalicylic acid with different solid dosage forms excipients. J Therm Anal Calorim. 2013;112:403–19.
Wesolowski M, Rojek B, Piotroska J. Application of chemometrically processed thermogravimetric data for identification of baclofen–excipient interactions. J AOAC Int. 2012;95:691–8.
Newman AW, Byrn SR. Solid-state analysis of the active pharmaceutical ingredient in drug products. Dr Discov Today. 2003;8:898–905.
Cano-Chauca M, Stringheta PC, Ramos AS, Cal-Vidal J. Effect of the carriers on the microstructure of mango powder obtained by spray drying and its functional characterization. Innov Food Sci Emerg Technol. 2005;6:420–8.
Gallo L, Piña J, Bucalá V, Allemandi D, Rámirez-Rigo MV. Development of a modified-release hydrophilic matrix system of a plant extract based on co-spray-dried powders. Powder Technol. 2013;241:252–62.
Acknowledgements
This study was supported by the Fapesq, Propesq/UEPB, Capes and CNPq (Process No.: 562957/2010-3). The authors especially thank Professor Ivan Coelho Dantas (in memoriam) for indicating the plant to be studied in this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Fernandes, F.H.A., de Almeida, V.E., de Medeiros, F.D. et al. Evaluation of compatibility between Schinopsis brasiliensis Engler extract and pharmaceutical excipients using analytical techniques associated with chemometric tools. J Therm Anal Calorim 123, 2531–2542 (2016). https://doi.org/10.1007/s10973-016-5241-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-016-5241-0