
Abstract
Ximenia americana L. is a plant of the Olacaceae family, used in the treatment of infectious diseases. Dry extracts are API’s of great interest for the pharmaceutical industry and have been used as final and intermediate products, resulting in different pharmaceutical forms. The aim of this study is to evaluate the compatibility of dry extract of X. americana with pharmaceutical excipients used in the solid forms. The extract was obtained by spray dryer, and binary mixtures of the dry extract and pharmaceutical excipients were analyzed. The studies were obtained using thermal analysis, X-ray powder diffraction and optical microscopy. With the data obtained from DSC curves, matrices for hierarchical cluster analysis (HCA) were made. The X-ray diffraction analysis showed changes in the mixtures’ profile with corn starch, lactose and magnesium stearate. Thermal behavior of the mixtures showed incompatibilities between the extract and lactose, corn starch and magnesium stearate. This study shows the importance of using instrumental analytical techniques in the early stages of development of herbal medicine to ensure the effectiveness, safety and quality of the final product.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ximenia americana L. is a typical plant of the tropics, being found mainly in parts of Africa, India, New Zealand, Central America and South America [1]. It is widely used in the folk medicine of these regions, and its roots are used to treat fever, jaundice, headaches and mental illness and as an antiseptic [2]. Studies have found evidence of their antiviral, analgesic and antimicrobial properties [3,4,5,6,7,8,9] corroborating its popular use in combating infections.
Preliminary studies indicated that the ethanolic extract of X. americana has as its main constituents: tannins, flavonoids, alkaloids, saponins, anthraquinones, starch, glycosides and bitter principles [10]. In the composition of its essential oil, 33 constituents were identified, being 69.0% aromatics, 12.5% lipid compounds and 13.0% of terpenoids [2]. In the seed’s oil, some constituents were identified as unsaturated fatty acids and other essential fats, such as linoleic acid, linoleic acid and arachidonic acid [1]. Other authors indicate the strong presence of vitamin C [11]. Le et al. [12] isolation and identification of the cyanogenic glycoside (sambunigrin), gallic acid, gallotannins β-glucogalline, 1,6-digalloyl-β-glucopyranose, quercetin, quercitrin (quercetin-3-O-α-rhamnopyranoside), avicularin (quercetin-3-O-α-arabinofuranoside), quercetin-3-O-β-xylopyranoside, quercetin-3-O-(6″-galloyl)-β-glucopyranoside and kaempferol-3-O-(6″-galloyl)-β-glucopyranoside. And Brandão et al. [13] determined that gallic acid is the most common chemical compound of X. americana.
In the pharmaceutical industry, the use of suitable technological adjuvants, along with the technology of spray drying, represents one important step in the assurance of the adequate stability and quality of plant extracts [14]. The incorporation of these dry extracts in formulations is recommended because they are easily obtained, standardized and embeddable in dosage forms. It also has high chemical, physical–chemical and microbiological stability [15].
Preformulation studies were conducted in order to allow the detection of chemical changes in the formulation of a compound, even at very low levels [16]. For analysis of solids, the method should allow for a given property to be investigated. Most studies of this nature associate particle characterization techniques (such as x-ray diffraction) with molecular characterization techniques (such as infrared spectroscopy) [17].
In this stage of pharmacotechnical development, it is necessary to evaluate the compatibility between drugs and excipients. This is because interactions can affect the nature of the chemical composition and hence its safety and efficacy [18].
Thermal analysis has been an extensively used analytical technique to study the sample properties as a function of temperature. Some authors have shown the applications of different thermal analysis techniques in pharmaceutical technology and the study of the compatibility of drugs [19,20,21,22,23] and the standardization and characterization of dry extracts from medicinal plants [14, 24,25,26].
Thus, the aim of this study is to evaluate the compatibility of the dry extract of X. americana, with pharmaceutical excipients used in the formulation of solid forms by analytical techniques and assisted by hierarchical cluster analysis (HCA) as chemometric technique.
Materials and methods
Sample preparation
The bark of Ximenia americana L. (AMCA) was collected from the semiarid region in the State of Paraíba, Northeast of Brazil. The voucher specimen was prepared and identified at the Professor Jayme Coelho de Morais herbarium (PRU), Federal University of Paraiba, under the number EAN-100493. The herbal material was dried at 40 °C. The hydroalcoholic extract was obtained by maceration of powdered bark in water/ethanol (30:70 v/v) solvent system for 72 h and then dried by Spray Dryer (Labplant), with onset temperature at 140 °C and endset between 90 and 95 °C, under 3.0 mL min−1 flow rate. Colloidal silicon dioxide (Aerosil® 200) was used as a stabilizer for the preparation of dry extract.
Binary mixtures
The binary mixtures were prepared in different concentrations using dry extract (AMCA) and the following excipients: corn starch (CS), lactose (LA), microcrystalline cellulose 101 and 102 (MC 101 and 102), magnesium stearate (MS), polyvinyl pyrrolidone K-30 (PVP K-30), talc (TAL), colloidal silicon dioxide (CSD), sodium starch glycolate (SSG), pregelatinized starch (PGS) and sodium croscarmellose (CRO). The compatibility studies were obtained using binary mixtures at the following proportions (w:w): 1:1, 1:2, 1:4, 1:6, 1:8 and 1:10 for CS, MC 101, MC 102 and LA, and 1:1, 1:2 and 2:1 for TAL, MS, AER, SSG, PGS, CRO and PVP K-30.
Thermal analysis parameter
The DSC curves were obtained on a TA Instruments calorimeter, model DSC Q20, using aluminum crucibles with about 2 ± 0.1 mg of samples under nitrogen atmosphere, at the flow of 50 mL min−1. Rising temperature experiments were conducted in the temperature range from 25 to 400 °C and heating rate of 10 °C min−1. Indium (m.p. 156.6 °C) was used as a standard for equipment calibration. Data were analyzed using the software TA Instruments Universal Analysis 2000, 4.7A.
The thermogravimetric curves were obtained with a simultaneous thermobalance module TG/DTA, model Q600 (TA Instruments), using alumina crucibles with about 8 ± 0.1 mg under a nitrogen atmosphere at 50 mL min−1. Rising temperature experiments were conducted in the temperature range from 25 to 900 °C and 10 °C min−1 heating rate. The apparatus was calibrated with calcium oxalate monohydrate.
Chemometric analysis
HCA as unsupervised pattern recognition technique analysis was applied to the calorimetric data of extract, excipients and binary mixtures. For a better view of the relations among the extract and excipients, the data were non-preprocessed. The models were built using Statistica 6.0 software, and the dendrogram was conducted in single linkage mode as amalgamation rule and Euclidian distance.
Fourier-transformed infrared spectroscopy (FTIR)
This analysis was performed using a Spectrum 400 spectrometer (PerkinElmer®) FTIR/FTNIR Spectrometer. The spectra of the samples were plotted in the range between 4000 and 500 cm−1, with a resolution of 4 cm−1, and sampled in powder form.
X-ray powder diffraction (XRPD)
The XRPD analysis was carried out in a XRD 6000 diffractometer (Shimadzu) with adopted scanning from 10° to 50° (2° min−1) on the 2θ scale and Cu (Kα1) radiation. The equipment was operated on 40.0 kV and 30.0 mA. The data were plotted by means of the software Origin® version 8.1.
Results and discussion
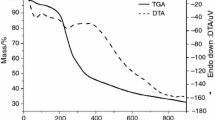
The DSC data of X. americana extract (Figs. 1a, 2a) demonstrate the presence of an endothermic peak at a temperature of 83.60 °C (∆H = − 270.50 J g−1), which is probably related to the loss of volatile constituents in the sample, such as ethanol and water. This event, shown in the thermogravimetric curve (Fig. 2a), corresponds to the loss of 7.92% of the sample mass. The decomposition process of the organic compounds starts at 157.11 °C and continues until the end of the analyzed range, representing a loss of 40.57% of the sample mass, resulting in a residue of 43.52%. This significant residue is probably due to the content of the drying adjuvant (20%), which increases the stability of the extract as it is very stable itself (m. p. 1600.0 °C) [27].

DSC curves: a—extract (AMCA); b—CS and its binary mixtures; c—LA and its binary mixtures; d—MC 101 and its binary mixtures; e—MC 102 and its binary mixtures; f—MS and its binary mixtures

TG curves: a—extract (AMCA); b—CS and its binary mixtures; c—LA and its binary mixtures; d—MC 101 and its binary mixtures; e—MC 102 and its binary mixtures; f—MS and its binary mixtures
According to the results, most of the extract–excipient mixtures exhibit no significant variations in the expected thermal profiles. Enthalpy values varied proportionally to the active substance content. Some changes concerning enthalpy are expected in such experiments due to problems in content uniformity of small samples used in DSC analysis [28].
The corn starch DSC profile exhibited its first endothermic peak at 150.79 °C, although an event related to the gelatinization process was not manifested. Differences in the thermal behavior of different corn starch lots are explained by the fact that starches from different plant sources differ in amylose/amylopectin ratios. These differences modify the physical properties of the starches, and the various types may not be interchangeable for a given pharmaceutical application [29]. The DSC analysis of binary mixtures between the extract and starch (Fig. 1b) demonstrates that the mixture assumes the characteristics of the excipient. Due to increasing the concentration of the starch in the study, the same event moves from 201.07 °C to the temperatures of 170.21, 176.21, 140.99, 143.92 and 211.49 °C, respectively, for 1:2, 1:4, 1:6, 1:8 and 1:10 mixtures. In this case, variations occur in the heat peaks, within a fairly wide range between 62.84 and 148.20 J g−1. Other characteristic processes of starch are also anticipated by the mixtures, but within a growth rate smaller than the event described above. In the DSC profiles, no events related to the extract are presented in any of the curves. This suggests incompatibility between the extract and the excipients in the proportions tested.
In the TG curve, it was observed that the binary mixtures with starch showed no change in the profile of the initial mass loss of the extract. In all curves, it is possible to identify the dehydration process of starch, up to about 100.00 °C, besides the main stage of mass loss between 256.06 and 338.91 °C, also characteristic of the excipient, followed by a slow degree of mass loss shown by the extract, which extends through the end of the curve. The sample’s mass lost at this stage (40.00–60.00%) is consistent with the amount of starch present in the sample (Fig. 2b).
The binary mixtures produced with lactose (Fig. 1c) expressed predominantly the characteristics of the excipient. The typical events of dehydration (147.84 °C), interconversion (217.47 °C) and decomposition (245.74 °C) are expressed by the lactose binary mixtures in the same temperature range. This profile characterizes the suppression of the extract characteristics, showing incompatibility between these components. Once there is no emergence of peaks in different temperature ranges, it will be possible to infer that this interaction is not chemical [30]. Due to the agglutinative function of lactose, the observed effect may indicate that this component can damage the liberation of the extract in the fluids of an organism, causing undesirable effects in the functionality of the formulation. Figure 2c shows changes in the properties of the extract, being the predominantly observed events of the excipient. Between 1:2 and 1:10 mixtures, a clear suppression of the first step of mass loss of the extract is visible, whereas it is more intense in the dehydration stage of lactose, which occurs between 129.01 and 152.22 °C. An important step in the decomposition of the extract, which occurs between 218.13 and 318.66 °C, is suppressed in the first mixture resulting in the manifestation of only the steps of the excipient degradation, occurring in two steps (the first between 211.62 and 252.32 °C and the second from approximately 252.32 °C). These thermogravimetric data confirm the incompatibility inferred by the DSC data, due to the distortion of the normal degradation profile of the extract. Interactions between other API’s and lactose are frequently reported in the literature [31,32,33].
The MC 101 (Fig. 1d) did not interact with the extract. This was based on the thermal profile of the mixtures, which have retained the characteristics of the extract, while expressing the endothermic peak of the excipient. For these mixtures, both the first (between 68.00 and 75.00 °C) and the second endothermic peaks (around 357.00 °C) of the excipient are expressed at the same temperature with no significant change in the height or the onset temperature of the event. Figure 2d shows that there was no change in the water loss during the first stage of the extract and excipient. It was detected in the mixtures that the main stage of mass loss of cellulose has its onset within normal temperature range (from 252.37 °C), being only slightly shifted to higher temperatures, and extending up to 372.79 °C. The percentage of mass lost in this step or the residue of the sample (ranging between 40 and 70%) is consistent with the amount of excipient contained.
In the MC 102 and its mixtures (Fig. 1e) the first peak, characteristic of loss of water, was expressed in temperature between 62.00 to 74.00 °C, while the second event is expressed between 352.00 and 360.00 °C with no significant change in the onset temperature of the events. In both cases, MC 101 and 102, the profiles show desirable compatibility, which indicate that the combination of the extract with these components may not modify its characteristics, while these diluents will perform an essential function in the solid formulation. The thermogravimetric curves obtained from mixtures with microcrystalline cellulose 102 (Fig. 2e) showed characteristics of both components. The 1:1 mixture expresses a characteristic mass loss of the extract in the temperature range between 30.00 and 126.49 °C. It was observed that the decomposition of the samples began to be recorded near the range 169.53–241.27 °C, resulting in a mass loss of 40–60%. As the concentration of excipient in the mixture was increased, the greater percentage of the sample was degraded in the range of decomposition of cellulose, between 238.40 and 388.57 °C. The percentage of residue at 900 °C ranged between 33.71 and 15.10% between 1:1 and 1:10 mixtures. Once no significant shifts were detected in the temperature range of events of both components, the thermal compatibility between the extract and microcrystalline cellulose 102 can be restated without compromising the thermal stability of these components when combined.
The magnesium stearate mixtures (Fig. 1f) showed that in the ratios of 1:1 and 1:2, the excipient influences the characteristics of the extract, while in the proportion of 2:1, the intensity of this interaction is reduced. This excipient is widely related to incompatibilities [18, 33], with various substances. However, in a solid formulation, it is used at a low concentration, causing interaction, but at a negligible magnitude. Probably, the products of the interaction in this ratio of components are not capable of causing significant change in the profile of the extract. Thermogravimetric data of magnesium stearate mixtures (Fig. 2f) showed different behaviors according to the variation in the proportion of the components. In the 1:1 mixture, it was possible to view characteristic events of both components. The first mass loss of the extract and excipient occurred between 30.00 and 104.49 °C. In the 1:2 mixtures, we can notice a mass loss that begins at a temperature of 157.09 °C, probably due to an event of the extract, since the carrier is stable at the same temperature. The combination 2:1 profile showed characteristic events of the extract with the same mass loss temperatures of this event. These data show that the interaction of the extract with ME occurs as a function of the excipient concentration in the formulation, reinforcing earlier observations. Therefore, once this excipient is used at low concentrations, the interaction can be reduced at negligible levels, without loss of its function.
The excipient PVP K-30 (Fig. 3a) displayed thermal compatibility with the extract, once it had stopped detecting changes in their thermal profile except for a slight anticipation of the event of loss of water from the mixture, which now occurs at about 75.00 °C. In Fig. 4a, there were no observable changes in the events that suggested compromising of the extract’s stability. The mass loss in the temperature range from 389.22 to 460.20 °C, characteristic of the excipient degradation, has not changed, nor the loss of water. The percentage of mass loss and residue between 13.52 and 21.34% is consistent with the proportion of the components in the mixtures. The conservation of the extract’s stability and its degradation profile confirm the compatibility between PVP K-30 and the extract, indicating a possible agglutinant which can be used in the solid formulation.

DSC curves: a—PVP K-30 and its binary mixtures; b—TAL and its binary mixtures; c—CSD and its binary mixtures; d—SSG and its binary mixtures; e—PGS and its binary mixtures; f—CRO its binary mixtures

TG curves: a—PVP K-30 and its binary mixtures; b—TAL and its binary mixtures; c—CSD and its binary mixtures; d—SSG and its binary mixtures; e—PGS and its binary mixtures; f—CRO its binary mixtures
Possible incompatibilities between the binary mixtures produced by the extract and talc were not observed (Fig. 3b). In Fig. 4b, mixtures with talc also showed mass loss in temperature ranges related to events of the extract (30.00–110.65 °C) and the beginning of the main stage of mass loss, near 138.92 °C. Shifts were not detected during any of the events. The proportion of waste was also proportional to the concentration of the components (between 58.96 to 77.13% for 2:1 and 1:2). The non-detection of distortions in the degradation profiles confirms the compatibility between talc and the X. americana extract.
The absence of alterations in the thermal profile of the extract when it was included in binary mixtures with CSD (Fig. 3c) is evidence of compatibility of the extract with this excipient. At all proportions studied, a characteristic endothermic event of the extract is identifiable at temperatures between 30.00 and 120.00 °C. Similarly, the TG curves of the binary mixtures (Fig. 4c) showed in its profiles all characteristic events of the extract with no stability loss of this component.
The DSC profiles of the binary mixtures with sodium starch glycolate (Fig. 3d) showed characteristic events of both components. In these profiles, it is possible to identify the endothermic event of moisture loss in the two components (both amorphous) at temperatures between 30.00 and 164.00 °C and the thermal decomposition of the excipient, an exothermic event between 263.36 and 364.24 °C. The events are also shown in the TG curves (Fig. 4d), in which is possible to notice that the decomposition events occur at the same temperature intervals with only differences in the mass loss at each event due to the different proportions of the samples. However, the thermal profile of the 1:1 mixture presented a decomposition event between 333.16 and 634.70 °C with greater mass loss by the sample (38.26%). This event is probably attributed to an anticipation of the extract decomposition, once the final residue of the sample is of 10.09%, when the 1:2 and 2:1 mixtures presented residues of 29.32 and 40.27%, respectively. The unexpected behavior of the 1:1 mixture provided evidence of incompatibility between the extract and sodium starch glycolate.
In Fig. 3e, the DSC profiles of the binary mixtures with pregelatinized starch show the suppression of the thermal events between 141.54 and 177.68 °C, characteristic of the beginning of the excipient decomposition. This event is not observed in any mixture studied [34, 35]. In the TG curves of the mixtures (Fig. 4e), there are no signs of stability loss of the components. Incompatibility with this excipient is reported in some studies, but the origin of the incompatibility is uncertain and probably related to the nature of the involved API in the binary mixture.
The DSC curves of the binary mixtures with sodium croscarmellose (Fig. 3f) showed characteristic events of the extract (moisture loss at 30.00–148.63 °C) and the excipient (thermal decomposition at 229.61–340.08 °C) with no displacement in the characteristic temperature intervals. In the TG curves (Fig. 4f), it is shown that the decomposition events of the samples occurred at the same temperature intervals (182.85–328.43 °C and 316.14–619.58 °C), corresponding to excipient and extract decomposition, respectively, and indicating no stability loss of these components.
Chemometric analysis
The thermal analysis allows to obtain a great quantity of data, and these data are often not available directly. Thus, it is necessary that the data treatment uses mathematical and statistical procedures, such as chemometric methods. The interaction between extracts and excipients can be better observed using pattern recognition techniques that present results as graphs [36]. To build the HCA dendograms, all data were used, including not only peaks but also other parts of the DSC curves that don’t contain thermal events. Figure 5 shows the HCA dendograms of the extract, excipients and mixtures of AMCA.

Hierarchical cluster analysis (HCA): a—CS and its binary mixtures; b—MC 101 and its binary mixtures; c—MC 102 and its binary mixtures; d—LA and its binary mixtures; e—PGCS and its binary mixtures; f—SSG and its binary mixtures
In the dendogram of mixtures with starch and extract (Fig. 5a), it was not possible to verify the formation of groups, and all mixtures were near excipient. This result shows that the starch profile was dominant over the extract, even at low concentrations, suggesting that starch interacts with the extract in these mixtures.
In the mixtures with microcrystalline celluloses 101 (Fig. 5b) and 102 (Fig. 5c), the resulting dendograms were very similar. With the cellulose, the formation of two major groups occurs separating the mixtures from pure extract and pure excipient, indicating the differentiation of the mixture’s profile. This can be attributed to the fact that all mixtures showed a characteristic thermal profile, resulting from the presentation of peaks of both the extract and of the excipient.
For lactose (Fig. 5d), the mixtures 1:1, 1:6 and 1:10 were distributed closer to the extract. The mixture 1:8 was between extract and lactose, indicating the interaction. The mixtures 1:2 and 1:4 were closer to lactose, indicating that the excipient contributes strongly in the DSC curves. The lack of a logical distribution of the mixtures in relation to the excipient indicates the absence of a pattern in the DSC curves, which suggests the interaction between these components.
The pregelatinized starch (Fig. 5e), sodium starch glycolate (Fig. 5f), magnesium stearate (Fig. 6a) and mixtures with AMCA demonstrate that the excipient was closer to one of the mixtures. This indicates the prevalence of the excipient profile in the mixture, indicating the interaction between excipient and extract.

Hierarchical cluster analysis (HCA): a—MS and its binary mixtures; b—PVP K-30 and its binary mixtures; c—CRO and its binary mixtures; d—CSD and its binary mixtures; e—TAL and its binary mixtures
The corresponding dendograms of the mixtures with PVP K-30 (Fig. 6b), sodium croscarmellose (Fig. 6c) and colloidal silicon dioxide (Fig. 6d) were similar to those of CMC 101 and CMC 102, with the separation of the mixtures from the pure extract and pure excipient, which indicates that the mixtures showed unique thermal profiles due to the association of characteristics from the DSC profiles of the extract and the excipient.
The mixtures with talc (Fig. 6e) behaved similarly to those of CMC-101, with the separation of the mixtures of the pure extract and pure excipient, except for the mixture 2:1, which was further from all DSC curves.
FTIR
In the FTIR spectrum related to a nebulized extract, a broad band is presented between 3800 and 3000 cm−1 which is indicative of hydroxyl groups (O–H stretch) [37]; those groups are attributed both to hydroxylated molecules of the extract (such as polyphenols) and to the moisture content of the extract, due to its amorphous nature. The peaks in the range from 2974 to 2847 cm−1, characteristic of carbon–hydrogen bonds [37], are attributed to the nature of the organic compounds contained in the extract. The sharp peaks in the region of 1702–1685 cm−1, relative to carbon–oxygen bonds (C=O) [38] as present in ethers, esters and carboxylic acids, are indicative of a variety of metabolites, such as tannins, flavonoids and anthraquinones, among others [39]. This range is also indicated by peaks in the regions 1307–1025 cm−1 (carbonyl), 1607–1516 cm−1 (C=C bonds in aromatic rings) and 788–674 cm−1 (substituted aromatic C-H bonds) [36].
In addition to the dry extract, the samples suspected of incompatibility were selected for analysis by FTIR. The spectrum of the binary mixture with corn starch (Fig. 7a) showed no alterations in the profile of the nebulized extract; the above-described extract bands were expressed in the spectrum, with superimposition of those relative to the excipient, suggesting no evidence of interactions between the functional groups of these components. Similar profiles were observed for the mixtures with sodium starch glycolate (Fig. 7d) and pregelatinized starch (Fig. 7e).

FTIR spectra: a—CS, b—LA, c—MS, d—SSG, e—PGS
The mixture with lactose showed, in its FTIR spectrum (Fig. 7b), absorption bands predominantly characteristic of the excipient, such as those in the regions of 3600–2700 cm−1 and 1500–800 cm−1. The extract characteristic bands at 1702–1685 cm−1 are expressed in the mixture spectrum, indicating a preservation of the carbonyl groups present in many compounds of the extract. However, the formation of a strong absorption band at the region below 800 cm−1 is indicative of chemical interactions between these components, which suggest incompatibility.
The FTIR spectrum of the binary mixture with magnesium stearate (Fig. 7c) occurred at 3000 cm−1. At the region of 3000–2700 cm−1, two sharp peaks are presented, which suggests the strong formation of terminal C-H groups. (The peaks may be due to its axial and angular deformations.) The suppression of the peak relative to the carbonyl group at the region of 1702–1685 cm−1 was also observed. These alterations are indicative of chemical interactions between the compounds of the extract and magnesium stearate, which allows for the classification of this mixture as incompatible.
XRPD
In order to prove possible extract crystallinity, the samples suspected of incompatibility were selected for analysis by X-ray diffraction (XRPD). The binary mixtures in the ratio 1:1 (w/w) were chosen for analysis in order to avoid any deviation on the diffraction profiles by the amount of the components. The diffraction profile of the dry extract was found to be predominantly amorphous, with three crystalline regions. This is probably due to the large number of compounds present in the amorphous phytocomplex, which is expected in plant extracts. The crystalline regions are attributed to the carbohydrates present in the extract, which may have crystallized during the drying process.
When combined with corn starch (Fig. 8a), the profile of the amorphous starch is expressed at the beginning of the curve with the expression of the crystalline regions of the extract at angles above 37.5°. The maintenance of both components’ structures in the mixture is evident that the interaction between the extract and starch detected on the thermal profiles is not of physical nature.

XRPD: a—CS, b—LA, c—MS, d—SSG, e—PGS
Lactose (Fig. 8b), being a disaccharide, has a stronger profile of crystallinity, which is mainly expressed in the mixture diffractogram. However, the characteristic crystalline peak of the extract above 42.5° is expressed in the mixture, which indicates conservation of the extract structure in the presence of this excipient.
In the mixtures with magnesium stearate (Fig. 8c), the profiles indicate certain amorphization of the sample, probably due to the amorphous phase of the extract, but also influenced by the conservation of the crystalline structures of the excipient and extract, which can be observed in the expression of the crystalline peaks at 37.5° and 42.5°.
Sodium starch glycolate (Fig. 8d) presents a crystallographic profile with multiple regions of crystallinity and amorphicity. These regions are expressed in the mixture profile, but those characteristics of the extract are suppressed in this sample, indicating an interaction between these components in solid state, which implies incompatibility.
The diffractographic profile of the mixture with pregelatinized (Fig. 8e) starch shows a completely amorphous profile, with suppression of the crystalline phase of the extract, which indicates a physical interaction between these components.
Conclusions
The use of instrumental analytical techniques is a valuable tool for the pharmaceutical industry. With some of these methods, it was possible to indicate excipients that may facilitate a solid formulation of the dry extract from X. americana, outlining a pharmaceutical form for this plant whose pharmacological activities have long been known by people from different regions of the world and proven by several studies.
The results show that lactose, starch and magnesium stearate showed incompatibility with the extract, causing considerable changes in the profile of diffraction and thermal properties with the suppression of the characteristics of the extract. Probably the inclusion of this excipient in the formulation would change the characteristics of bioequivalence and bioavailability of the extract, which is the criterion for the elimination of this excipient in the formulation.
This study also demonstrates the importance of using instrumental techniques in the early stages of development of herbal medicine, selecting excipients that can optimize the activity of the extract of medicinal X. americana.
References
Brasileiro MT, Egito AA, Lima JR, Randau KP, Pereira GC, Rolim Neto PJ. Ximenia americana L.: botany, chemistry and pharmacology in the pharmaceutical technology interest. Braz J Pharm. 2008;89:164–7.
Mevy J, Bessiere J, Greff S, Zombre G, Viano J. Composition of the volatile oil from the leaves of Ximenia americana L. Syst Ecol. 2006;34:549–53.
Asres K, Bucar F, Karting T, Witvrouw M, Pannecouque C, Clerq E. Antiviral activity against human immunodeficiency virus type 1 (HIV-1) and HIV-2 of ethnobotanically selected Ethiopian medicinal plants. Phytother Res. 2001;15:62–9.
Silva MSP, Brandão DO, Chaves TP, Formiga Filho ALN, Costa EMMB, Santos VL, Medeiros ACD. Study bioprospecting of medicinal plant extracts of the semiarid northeast: contribution to the control of oral microorganisms. Evidence Based Compl Alternat Med. 2012. doi:10.1155/2012/681207.
Silva KM, Chaves TP, Santos RL, Brandão DO, Fernandes FHA, Ramos Júnior FJ, Santos VL, Felismino DC, Medeiros ACD. Modulation of the erythromycin resistance in Staphylococcus aureus by ethanolic extracts of Ximenia americana L and Schinopsis brasiliensis Engl. Bol Lat Am Caribe Plan Med Aromat. 2015;14:92–8.
Soro TY, Traore F, Sakande J. Analgesic activity of the aqueous extract from Ximenia americana. CR Biologies. 2009;332:371–7.
James DB, Abu EA, Wurochekke AU, Orji GN. Phytochemical and antimicrobial investigation of the aqueous and methanolic extracts of Ximenia americana. J Med Sci. 2007;07:284.
Costa EMMB, Barbosa AS, Arruda TA, Oliveira PT, Dametto FR, Carvalho RA, Melo MD. In vitro antimicrobial activity of plant extracts against Enterococcus faecalis. Braz J Med Lab Path. 2010;46:175.
Omer MEFA, Elnima EI. Antimicrobial activity of Ximenia americana. Fitoter. 2003;74:122–6.
Ogunleye DS, Ibitoye SF. Studies of antimicrobial activity and chemical constituents of Ximenia americana. Trop J Pharm Res. 2003;02:239–41.
Silva GG, Souza PA, Morais PLD, Santos EC, Moura RD, Menezes JB. Wild plum fruit characterization (Ximenia americana L.). Braz J Frut. 2008;30:311–4.
Le NHT, Malterud KE, Diallo D, Paulsen SB, Nergård CS, Wangensteen H. Bioactive polyphenols in Ximenia americana and the traditional use among Malian healers. J Ethnopharmacol. 2012;139:858–62.
Brandão DO, Fernandes FHA, Ramos FJL, Silva PCD, Santana CP, Medeiros FD, Véras JG, Medeiros ACD. Validation of UPLC method for determination of gallic acid from Ximenia americana L. Planta Med. 2014. doi:10.1055/s-0034-1395050.
Medeiros ACD, Medeiros IA, Macêdo RO. Thermal studies of Albizia inopinata crude extract in the presence of cyclodextrin and Aerosil® by TG and DSC coupled to the photovisual system. Therm Acta. 2002;392:93–8.
Oliveira OW, Petrovick PR. Spray drying of plant extracts: basic remarks and application. Braz J Pharm. 2010;20:641.
Borchardt RT. Value of pharmaceutical sciences. J Pharm Biomed Anal. 1998;16:1387–401.
Chieng N, Rades T, Aaltonen J. An overview of recent studies on the analysis of pharmaceutical polymorphs. J Pharm Biomed Anal. 2011;55:618–44.
Tita B, Fulias A, Bandur G, Marian E, Tita D. Compatibility study between ketoprofen and pharmaceutical excipients used in solid dosage forms. J Pharm Biomed Anal. 2011;56:221–7.
Medeiros ACD, Cervantes NAB, Gomes APB, Macêdo RO. Thermal stability of prednisone drug and tablets. J Therm Anal Calorim. 2001;64:745.
Medeiros ACD, Correia LP, Simões MOS, Macêdo RO. Technological quality determination of pharmaceutical disintegrant by DSC cooling and DSC photovisual. J Therm Anal Calorim. 2007;88:311–5.
Santos AFO, Basílio Júnior ID, Souza FS, Medeiros AFD, Pinto MF, Santana DP, Macedo RO. Application of thermal analysis of binary mixtures with metformin. J Therm Anal Calorim. 2008;93:361–4.
Procópio JVV, Souza VG, Costa RA, Correia LP, Souza FS, Macêdo RO. Applicatin of thermal analysis and pyrolysis coupled to CG/MS in the qualification of simvastatin pharmaceutical raw material. J Therm Anal Calorim. 2011;106:665.
Oliveira AH, Moura EA, Pinto MF, Procópio JVV, Souza VG, Souza FS, Macêdo RO. Thermal characterization of raw material pentoxifylline using thermoanalytical techniques and Pyr-CG/MS. J Therm Anal Calorim. 2011;106:763–6.
Fernandes FHA, Santana CP, Santos RL, Correia LP, Conceição MM, Macêdo RO, Medeiros ACD. Thermal characterization of dried extract of medicinal plant by DSC and analytical techniques. J Therm Anal Calorim. 2013;113:443–7.
Aragão CFS, Souza FS, Barros ACS, Veras JWE, Barbosa Filho JM, Macêdo RO. Aplicação da termogravimetria (TG) no controle de qualidade da milona (Cissampelos sympodialis Eichl.) Menispermaceae. Braz J Pharmacog. 2002;12:60–1.
Correia LP, Procópio JVV, Santana CP, Santos AFO, Cavalcante HMMO, Macêdo RO. Characterization of herbal medicine with different particle sizes using pyrolysis GC/MS, SEM and thermal techniques. J Therm Anal Calorim. 2011;123:993–1001.
Rowe RC, Sheskey PJ, Quinn MC. Handbook of pharmaceutical excipients. 6th ed. Washington: Pharmaceutical Press; 2009.
Maximiano FP, Novack KM, Bahia MT, Sá-Barreto LL, Cunha-Filho MSS. Polymorphic screen and drug–excipient compatibility studies of the antichagasic benznidazole. J Therm Anal Calorim. 2011;106:819–24.
Souza FS, Barreto APG, Macêdo RO. Characterization of starch pharmaceuticals by DSC coupled to a photovisual system. J Therm Anal Calorim. 2001;64:739–43.
Desai SR, Shaikh MM, Dharwadkar SR. Preformulation compatibility studies of etamsylate and fluconazole drugs with lactose by DSC. J Therm Anal Calorim. 2003;71:651–8.
Cides LCS, Araújo AAS, Santos-Filho M, Matos JR. Thermal behaviour, compatibility study and decomposition kinetics of glimepiride under isothermal and non-isothermal conditions. J Therm Anal Calorim. 2006;84:441–5.
Verma RK, Garg S. Compatibility studies between Isosorbide Mononitrate and selected excipients used in the development of extended release formulations. J Pharm Biomed Anal. 2004;35:449–58.
Oliveira GGG, Ferraz HG, Matos JSR. Thermoanalytical study of glibenclamide and excipients. J Therm Anal Calorim. 2005;79:267.
Ibrahim HK, El-Setouhy DA. Valsartan orodispersible tablets: formulation, in vitro/in vivo characterization. AAPS Pharm Sci Tech. 2010;11:189–96.
Bharate SS, Bharate SB, Bajaj AN. Incompatibilities of pharmaceutical excipients with active pharmaceutical ingredients: a comprehensive review. J Excip Food Chem. 2010;1:3–26.
Fernandes FHA, Almeida VE, Medeiros FD, Silva PCD, Simões MOS, Véras G, Medeiros ACD. Evaluation of compatibility between Schinopsis brasiliensis Engler extract and pharmaceutical excipients using analytical techniques associated with chemometric tools. J Therm Anal Calorim. 2016;123:2531–42.
Skoog DA, Holler FJ, Nieman TA. Principles of Instrumental Analysis. 5th ed. Philadelphia: Harcourt Brace & Company; 1998.
Silverstein RM, Webster FX, Kiemle DJ. Identificação Espectrométrica de Compostos Orgânicos. 6th ed. Rio de Janeiro: Livros Técnicos e Científicos; 2006.
Júnior JOCS, Vieira JLF, Barbosa WLR, Pereira NL. Caracterização físico-química do extrato fluido e seco por nebulização de Symphytum officinale L. Braz J Farmacogn. 2006;16:671–7.
Acknowledgements
The authors thank to PROPESQ/UEPB, FAPESQ, CNPq (Grant No. 484408/2010-0) for financial supporting this research and CERTBIO/UEPB for analysis, and the authors especially thank to the Professor Ivan Coelho Dantas (in memoriam) for indicating the plant to studied in this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Santana, C.P., Fernandes, F.H.A., Brandão, D.O. et al. Compatibility study of dry extract of Ximenia americana L. and pharmaceutical excipients used in solid state. J Therm Anal Calorim 133, 603–617 (2018). https://doi.org/10.1007/s10973-017-6764-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-017-6764-8