
Abstract
The isobaric heat capacities of solid explosive materials were measured using differential scanning calorimeter. The values of C p were determined at temperature 298.15 K and compared to theoretical data calculated using the group additivity approach. Several group contribution methods were tested, showing that certain method for some explosives provides estimation of C p with error only 1 %. Generally, the average error limit for calculated C p was about 11 %, which is insufficient.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Explosives are chemical substances or mixtures of chemical substances in metastable state. Explosives can be used in a number of ways, including explosive cladding, metal working (forming, welding and cutting) and shearing by pyrotechnics systems. However, the principal non-military use of explosives is in mining, quarrying, tunnelling, dam building, demolition, construction and other applications [1, 2]. Their inherent property, which makes them different from other chemicals, is ability to rapidly release chemical energy after proper initiation. The most common type of initiating event is some sort of a heat impulse (heat, flame, spark, local adiabatic compression, etc.), and it is therefore important to determine thermal stability and sensitivity of explosives. The thermal stability is important in shelf life analysis and evaluation of storage requirements, while thermal sensitivity is crucial for determination of temperature limits for safe handling and processing. Variety of methods exists and is routinely in use for evaluation of thermochemical properties of explosives [1]. Measurement of isobaric heat capacity (C p) of explosives is not frequently published although this quantity can be used to calculate other thermodynamic properties. Moreover, C p value is necessary for evaluation of experiments done by using accelerating rate calorimetry whose experimental results lead to most realistic approximation of real sample storage scenario (more about accelerating rate calorimetry can be found in [3–5]). However, C p data are not so easily found in open literature.
The aim of this work is to determine the isobaric heat capacity values at 298.15 K for selected explosive materials in solid phase.
Experimental
The following explosive substances with purity over 95 % were selected: (a) standard explosives PETN, RDX, TNT, α-HMX, picric acid; (b) military explosives FOX-7, α-HNIW, ε-HNIW, NTO, A-IX-1, DINA, TNAZ, NQ, TEX; (c) improvised explosives DDNP (Dinol), UNi, ETN, TATP, DADP, HMTD; and (d) primary explosives MF (mercury fulminate), GNGT (Tetrazene), LA (lead azide), LA dextrinized.
The molar heat capacities were measured using DSC Pyris 1 (PerkinElmer) with intracooler flushed with dry nitrogen at flow rate 20 cm3 min−1. The temperature was calibrated to match melting point of several pure metals (Hg, Ga, In, Sn, Pb and Zn); the enthalpy was calibrated using enthalpy of fusion of indium. The metals necessary for calibration were weighted into aluminium pans. Explosive samples of mass about 10 mg were measured in sealed aluminium pans. The heat capacity measurements were repeated five times for each sample and standard material (sapphire, standard reference material 720, NIST) in the temperature range from (293 to 303) K with a heating rate 5 K min−1 with isotherm 2 minutes at the beginning at 293 K and 1 minute at the end at 303 K (optimal conditions found in previous work [6]). The evaluation of C p by so-called modified stepwise method is based on comparison between baseline, standard material and sample DSC curves and the areas under these curves, respectively (see Fig. 1). The heat capacity of studied sample can be calculated using Eq. (1), where C p is the isobaric specific heat capacity, m is a sample mass and A is the area under relevant curve. Subscript b stands for baseline, star in superscript is used for sapphire and the values without star are for the sample.

Heat flow dependence on time (a), or temperature (b) for baseline, sapphire and PETN for heat capacity determination by modified stepwise method. The temperature range for integration is emphasized, and the symbols of evaluation correspond to Eq. 1
In the case of standard stepwise method (described in Ref. [7, 8]), the entire area bellow the curve (from before and after isothermal step) is used for C p calculation. The modified stepwise method is based on integration of short temperature interval of the “step” [6]. The specific heat capacity of studied samples at the temperature 298.15 K was determined by the integration of each curve in the temperature range from 297.15 to 299.15 K as illustrated in Fig. 1b. Figure 2 shows DSC data of selected samples and sapphire as standard material. The deviation of heat capacity determined by DSC was tested using two “standard” materials (sapphire, standard reference material 720, NIST and molybdenum, reference material 781D2, NIST). The results showed deviation of C p value under 2 % [6].

Dependence of heat flow on temperature for selected samples
To make sure that the DSC will not be endangered in tested temperature range, all samples were first evaluated by explosion proof differential thermal analysis in DTA Ex550 (OZM Research). The decomposition temperatures were determined for all samples prior to heat capacity measurements. The calibration of DTA was done using indium (melting point). The sample mass varied based on the type of substance measured. Thermal decomposition of primary explosives is quite violent, and they were therefore tested in amount of 5 mg. Less violently decomposing secondary explosives were measured in amount of 50 mg. All samples were measured at linear heating rate 5 K min−1 in glass micro-test tubes under static air atmosphere.
Results and discussion
Explosive materials included in this study were selected to represent major types of explosives including both primary and secondary explosives. Commercially available materials and improvised explosives are included. Heat capacities (C p) are available for some of the commercially available secondary explosives, and those were used for verification of our results. Data presented in this article for primary and improvised explosives are, to our best knowledge, not available in open literature.
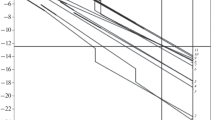
DTA curves were used to characterize all the samples. The endothermic peaks were phase transition such as melting of the sample; the exothermic effects were always decompositions. The illustration of obtained DTA signals is given in Fig. 3. The temperatures of all endothermal and exothermal processes are summarized in Table 1, where symbol S stands for start and O for onset of the effect. In the cases of RDX, A-IX-1, TEX and UNi, endothermic effects overlap with exothermic one; thus, only the temperature of the maximum (max) of exothermic effect is given.

Temperature dependence of DTA signals of selected samples
The isobaric heat capacity was measured by modified stepwise method using DSC. Values of the specific heat capacity calculated according Eq. 1 are summarized in Table 1 for all studied samples alongside with the literature data. The experimental results obtained by authors are in good agreement with published values. The published values were in some cases obtained under different experimental conditions and therefore are not directly comparable with our results (data with note in Table 1).
Mercury fulminate (MF) and lead azide (LA) exhibit the lowest values of C p (see Table 1). This may be due to their inorganic-like structure. Higher values of C p but still below 1 J g−1 K−1 were determined for TEX and both forms of HNIW, which are substances with complicated caged structure. The rest of the samples are aliphatic substances and heterocycles without any systematic difference in C p. Measured values were for all of the samples in these two groups close to 1.1 J g−1 K−1. The highest value of C p was obtained for the two organic peroxides—DADP and TATP. This may be related to the high oxygen content in their molecules.
The precise experimental measurement of C p is quite time-consuming and experimentally demanding [19]. Methodologies enabling estimation of C p with reasonable accuracy were therefore proposed in the past. Generally, the methods can be divided into two groups; the first one is based on quantum–mechanical calculations and the second one on composition/structure of the substance (group contribution methods).
Sallamie and Shaw [20] developed a predictive technique employing density functional theory combined with the Debye–Einstein model to compute solid state C p. It should be noted that this method is computationally intensive [20, 21]. On the contrary, the group contribution methods are easy to use, and the numerical treatment is very simple. These methods are used for estimation of the heat capacities in this paper. It should, however, be noted that these methods suffer from a limited number of group values that have been evaluated.
The group contribution methods assume that various groups (or only elements) of the molecule contribute to the total molar heat capacity. The application of majority of these methods is limited to narrow temperature ranges, and in most cases, they have been designed for specific groups of organic compounds (e.g. very precise method by Domalski and Hearing [22] is only for hydrocarbons at 298.15 K).
The simplest method is based on element contribution to the heat capacity of the substance. Kopp [23] presented values for fundamental elements of organic compounds but without value for nitrogen. Thus, only C p value for TATP and DADP can be calculated by this method. The value calculated for both substances (1.628 J g−1 K−1) is higher than experimental results.
Hurst and Harrison [23] modified Kopp’s rule to be able to estimate the solid and liquid heat capacities at temperature of 298 K. They tested their method and declared that the average absolute errors for the correlations are 9.6 % for solid. The values calculated for substances studied in this work are summarized in Table 2. It can be seen that the values are within 10 % error from experimental results for 12 substances, from 20 to 25 % for 5 substances and over this limit for 2 substances. There does not seem to be correlation between the structure of the substance and the level of error of estimated C p. Although the highest errors were estimated for substances with complicated cyclic structure where nitrogen or oxygen is situated into the cycle, there are exceptions—TNAZ with error of only 1.2 % where nitrogen is in the cycle or DADP with error of 0.8 % where oxygen is in the cycle. The estimations for aromatic substances (TNT, picric acid, DINOL A) exhibit low error limits. Generally, the average error limit of all estimated C p values is 11.8 % using Hurst and Harrison method.
Rihani and Doraiswamy [24] published group contributions method with ring formation corrections (their method is based on data for cycloalkane and alkene rings). Unfortunately, the published groups correspond to simpler structures without contributions of oxygen and nitrogen. The C p values calculated by this method could therefore be determined only for 5 of the studied samples—TNT, picric acid (aromatic) and PETN, ETN, DINA (aliphatic). The estimated C p values are lower than the experimental ones, and the difference is higher for aromatic substances than for aliphatic. The values of C p for other substances as given in Table 3 are only rough estimation with simplification (e.g. correction to 6-membered ring instead of 8-membered for α-HMX), and obtained values are in all cases lower then experimental results. The Rihani and Doraiswamy method can provide only rough estimation of C p value with the average error limit of 18.6 % for our type of substances.
Krevelen and Nijenhuis [25] in their work emphasized contribution method developed by Satoh [26] bases on group contributions without corrections for ring formation. The limiting for his method is the absence of values for some important groups: –COO–, –CONH–, –SO2– and –F. Nevertheless, Satoh’s method allows calculation of C p for majority of substances studied in this work (see Table 3). The agreement with experimental values is better for Satoh’s method and then for method proposed by Rihani and Doraiswamy, but the average error is 12.8 %.
Chickos et al. [27] published the group contribution method for heat capacity estimation of the condensed phase substances at 298 K with accuracy within 26.9 J mol−1 K−1 on data set containing 446 solids. The authors claimed that the standard error of estimation using their approach is almost the same as was a typical experimental uncertainty of their studied solids. The calculated values of C p for studied samples as summarized in Table 2 give the average error of 13.2 %, but two substances, PETN and ETN, have error close to 50 %, and the calculated values are in both cases higher than the experimental ones. These results show that the error is in general higher than the Chickos et al. [27] declared in their work.
Goodman et al. [28] presented two methods for the estimation of C p of solid organic compounds applicable to two temperature regions higher than 50 K. The power-law method utilizes empirical temperature dependence, while the partition function method is based on the Einstein–Debye partition function for crystals with a modified frequency distribution function. Both methods have fixed temperature functionality but utilize group contributions to obtain the compound-specific constants in the predictive equations. Tests on the methods’ extrapolation capabilities suggest that at 298 K they have comparable accuracy to Domalski and Hearing [22] method or method of Hurst and Harrison [23]. The power-law method was used to calculate the C p values for studied samples, and the results are summarized in Table 2. This method provides results without any extreme value as was obtained in previous case by method of Chickos et al. [27], and the average error is 11.2 %.
The comparison of all methods for estimation of C p leads us to conclusion that the most accurate results were obtained using Hurst and Harrison [23] method and Goodman et al. [28] method.
Conclusions
The isobaric heat capacity of selected explosive materials was measured using differential scanning calorimeter. The heat capacity at 298.15 K was determined for 24 explosive materials in solid state, where for 16 of them the heat capacity data were not presented in open literature up to now. Several group contribution methods were used to estimate C p values for studied samples. The comparison with experimental results shows that the average error limit of estimated C p is not lower than 11 %, which is insufficient when heat capacity data should be used for other calculations.
References
Boileau J, Fauquignon C, Hueber B, Meyer HH. Explosives. In: Ullmann´s, editor. Encyclopedia of industrial chemistry. England: John Wiley and Sons; 2009.
Dobratz BM. LLNL explosives handbook. UCRL Report 52 997. Change 2. Livermore: ISEE EXPLOSIVES ENGINEERS; 1985.
Bohn MA, Volk F. Adiabatische Selbstaufheizung bei Treib-und Explosivstoffen. 24th Annual Conference of ICT, Karlsruhe; Jun 1993.
Accelerating rate calorimeter—ARC technical information note 005, temperature of no return: Application of ARC data maximum safe temperatures and pack sizes. Technical manual, THT.
Fischer HG, Goezt DD. Determination of self-accelerating decomposition temperatures using the Accelerating Rate Calorimeter. J Loss Prev Process Ind. 1991;4:95–9.
Pilař R, Honcová P, Koštál P, Sádovská G, Svoboda L. Modified stepwise method for determining heat capacity by DSC. J Therm Anal Calorim. 2014;118:485–91.
Mraw SC, Naas DF. Measurement of accurate heat-capacities by differential scanning calorimetry. Comparison of d.s.c. results on pyrite (100 to 800 K) with literature values from precision adiabatic calorimetry. J Chem Termodyn. 1979;11:567–84.
Application note M149-v1, Setaram.
Meyer R, Kohler J, Homburg A. Explosive. 5th ed. Wiley: Weinheim; 2002.
Baytos JF. Informal report LA-8034-MS, UC-45. Los Alamos Scientific Laboratory: University of California; 1979.
Haishan D, Fenfen Z. Performance of high explosives and their related materials. Beijing: Science Press; 1989.
Tran TD, Simpson RL, Maienschein J, Tarver C. U.S. Department of Energy. Karlsruhe, Germany; 3–6 Jul 2001.
Rylance J, Stubley D. Heat capacities and phase transitions of octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX). Thermochim Acta. 1975;13:253–9.
Licht HH, Hansson J. HMX (octogen) and its polymorphic forms. In: Symposium on chemistry problems with the stability of explosives. 1971. 3. p. 168–79.
Lyman JL, Liau YC, Brand HV. Thermochemical functions for gas-phase, 1,3,5,7-tetranitro-1,3,5,7-tetraazacyclooctane (HMX), its condensed phases, and its larger reaction products. Combust Flame. 2002;130:185–203.
Xu K, Song J, Zhao F, Ma H, Gao H, Chang C, Ren Y, Hu R. Thermal behaviour, specific heat capacity and adiabatic time-to-explosion of G(FOX-7). J Hazard Mater. 2008;158:333–9.
Zhaona L, Haixia M, Jirong S, Fengqi Z, Kangzhen X, Rongzu H. Specific heat capacity, thermodynamic properties and adiabatic time-to- explosion of 3-nitro-1,2,4-triazol-5-one (NTO). Huozhayao Xuebao. 2008;31:25–8.
Dorofeeva OV, Tolmach PI. Estimation of the thermodynamic properties of nitroguanidine, hexahydro-1,3,5-trinitro-1,3,5-triazine and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine in the gas phase. Termochim Acta. 1994;240:47–66.
Fulem M, Laštovka V, Straka M, Růžička K, Shaw JM. Heat capacities of tetracene and pentacene. J Chem Eng Data. 2008;53:2175–81.
Sallamie N, Shaw JM. Heat capacity prediction for polynuclear aromatic solids using vibration spectra. Fluid Ph Equilib. 2005;237:100–10.
Wunderlich B. Thermal analysis of polymeric materials. Heidelberg: Springer; 2005. p. 101–44.
Domalski ES, Hearing ED. Estimation of the thermodynamic properties of hydrocarbons at 298.15 K. J Phys Chem Ref Data. 1988;17:1637–78.
Hurst JE, Harrison BK. Estimation of liquid and solid heat capacities using a modified Kopp’s rule. Chem Eng Commun. 1992;112:21–30.
Rihani DN, Doraiswamy LK. Estimation of heat capacity of organic compounds from group contributions. Ind Eng Chem Fundam. 1965;4:17–21.
van Krevelen DW, te Nijenhuis K. Properties of polymers, their correlation with chemical structure; their numerical estimation and prediction from additive group contributions. 4th ed. Amsterdam: Elsevier; 2009.
Satoh S. Heat capacity and chemical constitution. J Sci Res Inst (Tokyo). 1948;43:61–93.
Chickos JS, Hesse DG, Liebman JF. A group additivity approach for the estimation of heat capacities of organic liquids and solids at 298 K. Struct Chem. 1993;4:261–9.
Goodman BT, Wilding WV, Oscarson JL, Rowley RL. Use of the DIPPR database for development of quantitative structure—Property relationship correlations: heat capacity of solid organic compounds. J Chem Eng Data. 2004;49:24–31.
Acknowledgements
This work was done under the financial support of Ministry of Interior of the Czech Republic, project no. VG20102014032. Authors thanks to Monika Subrtova for her help with DTA measurements.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Pilar, R., Pachman, J., Matyáš, R. et al. Comparison of heat capacity of solid explosives by DSC and group contribution methods. J Therm Anal Calorim 121, 683–689 (2015). https://doi.org/10.1007/s10973-015-4611-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4611-3