
Abstract
Microcalorimetry is a highly sensitive experimental technique that allows to determine the energy released by any process or transformation. In the field of medicine, it is interesting for investigations of microbial processes. The interaction relationship between Enterococcus faecalis and Pseudomonas aeruginosa was researched using a Tian–Calvet calorimeter equipped with two stainless steel cells (reference and experimental). Three samples from both bacteria were prepared in the following proportions: 20 + 80 % (0.2 mL E. faecalis + 0.8 mL P. aeruginosa), 50 + 50 % (0.5 mL E. faecalis + 0.5 mL P. aeruginosa) and 80 + 20 % (0.8 mL E. faecalis + 0.2 mL P. aeruginosa). Experiments were carried out at a concentration of 103 CFU mL−1 and a constant temperature of 309.65 K. Recording the heat voltage difference versus time, the growth curves for E. faecalis, P. aeruginosa and their mixtures were obtained. The differences in shape of curves of single microorganisms and their mixtures were compared. Also, the thermokinetic parameters of single microorganisms and their mixtures (growth constant, generation time, detection time and amount of heat released) were calculated.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Microcalorimetry is an analytical technique which allowing measurement of heat flow related to biological processes, which is proportional to the rate at which a given chemical or physical process takes place [1]. It is a nondestructive method, with high sensitivity, accuracy and simplicity, which has been extensively applied in physics, chemistry, life sciences and other fields. Also, it provides us some qualitative information, such as the real-time thermogenic fingerprints (heat flow–time curves), and many quantitative parameters, such as growth constant (k), the maximum thermal power (V max), the appearance time of maximum thermal power (t max), the generation time (G) and the amount of heat released (Q). Knowledge of these parameters facilitates the identification and study of the growth of bacterial species. However, this technique presents drawbacks, which are its lack of specificity and that it requires an initial equilibration time of approximately 2 h [1–3].
Microcalorimetry arouses interest in the biological sciences for its potential application in the study of the microbial activities in vitro [4]. Microorganisms produce small amounts of heat in a range of 1–3 pW per cell. Despite the low bacterial heat, their exponential growth in culture media permits their detection within a few hours, even from samples with a low concentration, e.g., 10 colony forming units (CFU) mL−1 [3, 5]. In the medical field, this method is very interesting due to the fast-growing nature of microorganisms and the resultant amount of heat that is soon produced [4]. In the last few years, it has been used to study the rapid detection of bacterial growth under different conditions [6–9] or microbial contamination [10–12]. Also, it has been employed to determine inhibitory effects and/or the minimal inhibitory concentration for different antimicrobial substances [13–16] and antibiotics [17–19] in a few hours. Similarly, antibacterial coatings for implant materials have also been tested using this technique [20]. In addition, microcalorimetry permits rapid differentiation between two species of bacteria of the same genus [21] and rapid detection of resistant strains of a bacterial species within hours [22, 23].
However, despite all the references mentioned above, there are few studies that attempt to investigate the mechanism of interaction between bacteria of clinical relevance employing microcalorimetric techniques [24, 25]. In our study, we researched the interaction relationship between two pathogenic bacteria, Enterococcus faecalis and Pseudomonas aeruginosa.
E. faecalis is a Gram-positive cocci that grows in pairs or chains and ferment carbon sources to produce lactid acid. It is a commensal organism in the gastrointestinal tract of healthy individuals as well as many other animals. This bacterium can act as opportunistic pathogens in compromised patients with nosocomial infections. E. faecalis is intrinsically resistant to many antibiotics and often requires synergistic antibiotic combinations to successfully treat infection [26].
P. aeruginosa is a nonfermenting, aerobic, motile Gram-negative rod that is ubiquitous in the natural environment. It has minimal nutritional requirements and can easily contaminate wet surfaces in the hospital, as well as equipment, devices and solutions. The bacterium secretes a biofilm that protects it and allows it to adhere to tissues and expresses a wide range of virulence factors. P. aeruginosa is an important opportunistic pathogen that causes a variety of infections in immunocompromised patients and hospitalized patients. Also, it is resistant to many commonly used antibiotics [26].
Materials and methods
E. faecalis (ATCC 29212) and P. aeruginosa (ATCC 27853) were provided by American Type Culture Collection (ATCC). These bacterial strains were streaked onto blood agar plates and incubated at 309.65 K for 24 h. The blood agar plates with multiple bacterial colonies were then used to prepare a bacterial suspension with sterile saline (0.9 % NaCl) and adjusted to an optical density of 0.5 on the McFarland scale. This suspension was diluted with 0.9 % sterile saline to obtain final concentration of 103 CFU mL−1. The inoculum concentration was confirmed using the plate count method. Later, mixtures of both bacteria were prepared in the following proportions: 20 + 80 % (0.2 mL E. faecalis + 0.8 mL P. aeruginosa), 50 + 50 % (0.5 mL E. faecalis + 0.5 mL P. aeruginosa) and 80 + 20 % (0.8 mL E. faecalis + 0.2 mL P. aeruginosa).
The measures were carried out using Tian–Calvet microcalorimeter equipment designed by Professor Paz Andrade [27]. It is equipped with a device allowing operation in the absence of vapor phase and has two Teflon® screw-capped stainless steel cells of approximately 10 cm3 (Fig. 1). One of these cells contains the reference solution, named the reference cell, and the other one the sample, named the experimental cell. A Philips PM2535 multimeter and a data acquisition system were linked to the microcalorimeter. Calibration was performed electrically using a Setaram EJP30 stabilized current source. The precision in calorimetric signal was ±0.01 mW. Further details about the experimental method have been already published by [3, 28].

Scheme of the experimental cell of Tian–Calvet microcalorimeter
The external media of the calorimeter was maintained at a constant temperature of 309.65 K. The reference cell was filled with 7 mL of culture medium +1 mL of sterile saline, while the experimental cell was injected with 7 mL of culture medium. The culture medium used was a liquid enriched with soybean-casein (Becton, Dickinson and Company, USA), which is a mixture of processed water (40 mL), soybean-casein digest broth (2.75 % w/v), yeast extract (0.25 % w/v), animal tissue digest (0.10 % w/v), sodium pyruvate (0.10 % w/v), dextrose (0.06 % w/v), sucrose (0.08 % w/v), hemin (0.0005 % w/v), menadione (0.00005 % w/v), sodium polyanetholsulfonate (0.020 % w/v) and pyridoxal HCl (0.001 % w/v). Both cells were introduced from the upper part of the calorimeter in the internal thermopile chamber through two cylindrical holes aligned in parallel. The large distance that separates the cells from the entrance ensures the minimization of heat flow to the exterior. The system is then allowed to stabilize during 2 h, after which 1 mL of the sample is introduced into the experimental cell.
The experiment was also carried out with a sample not containing any bacteria (control). All experiments were realized in triplicate.
Both cells were cleaned and sterilized by autoclaving before using (20 min at 394.15 K).
A data collection and processing system were used to record the electrical signal at intervals of 20 s throughout the duration of the experiment.
Results
When the suspensions of single microorganisms and their mixtures are introduced into the reference cell, the heat voltage signals are recorded to form exothermic heat flow–time curves of E. faecalis, P. aeruginosa and the mixed microorganisms at different proportions.
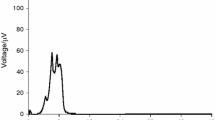
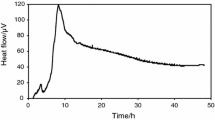
The curve of E. faecalis [29] (Fig. 2) had one single energetic phase with four peaks of heat flow, and the signal was recorded for about 10 h, returning to baseline levels after this time. The curve of P. aeruginosa [3] (Fig. 3) also presented one phase, where the ascending part showed four discrete peaks and the descending one an exponential shape that was extended in time. Probably, this last phase of the curve is related to the ability of this organism to form biofilms, allowing survival in the culture medium, without a net increase in cell number. Such behavior has been observed in various microorganisms that produce biofilms [30, 31]. Although E. faecalis also forms biofilm, P. areuginosa is much more efficient at making biofilms.

Graphical representation of heat flow versus time for the E. faecalis at 103 UFC mL−1

Graphical representation of heat flow versus time for the P. aeruginosa at 103 UFC mL−1
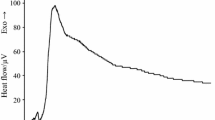
In the graphs of mixtures of E. faecalis and P. aeruginosa (Fig. 4), we have seen that the curve of 20 % E. faecalis + 80 % P. aeruginosa presented one phase with four peaks of thermal power separated by a stretch of the concave curve. The curve of 50 % E. faecalis + 50 % P. aeruginosa consisted in one phase with two peaks of voltage. In both cases, E. faecalis modifies the growth profile of P. aeruginosa, even at low proportions in the sample. Finally, in the graph of 80 % E. faecalis + 20 % P. aeruginosa, two metabolic phases were observed, the first with four lower energy peaks which signal was recorded for about 10 h, and is characteristic of the E. faecalis curve; and the second of greater energy that is prolonged over time and is similar to the curve of P. aeruginosa.

Graphical representation of heat flow versus time for E. faecalis and P. aeruginosa at 103 UFC mL−1 for the different mixtures studied: a 20 % E. faecalis + 80 % P. aeruginosa; b 50 % E. faecalis + 50 % P. aeruginosa and c 80 % E. faecalis + 20 % P. aeruginosa
The results were processed mathematically by adjusting them for exponential equations in ascending and descending phases of each peak of heat flow and polynomial equations in the areas of higher activity of the curve.
By means of the first derivative of the polynomial equations, we determine the value of the maximum thermal power (V max) and the time of its registration (t max) in the curves of E. faecalis, P. aeruginosa and their mixtures in different proportions. Although we cannot establish a quantitative proportionality relationship of the maximum peaks and their appearance time between pure cultures and their mixtures, all experiments show a maximum peak before 20 h (Table 1).
The detection time of the signal (t d) of E. faecalis, P. aeruginosa and their mixtures are presented in Table 1. As can be seen, the sample 20 % E. faecalis + 80 % P. aeruginosa has a t d higher than 50 % E. faecalis + 50 % P. aeruginosa and 80 % E. faecalis + 20 % P. aeruginosa. This is because E. faecalis has a t d lower than P. aeruginosa. In all curves, growth can be detected in less than 8 h.
In the logarithmic phase of the growth curve, we fitted the data to an exponential equation that can be expressed as [32, 33]:
where n 0 is the number of bacteria at time 0, n t the number of bacteria at time t and k the growth constant.
If P w is the thermal power of each cell, then:
Taking into account that P 0 and P t are the energies released at onset and at time t, respectively:
Thus, the growth constant (k) can be obtained by selecting two points from the exponential phase of a bacterial growth curve.
The generation time (G) is defined as the time taken by a population to duplicate its number and is expressed as:
Therefore, deriving from the mathematical adjustment of the powers obtained from the microcalorimetric study of bacterial cultures, we can quickly and easily extrapolate the value of the growth constant and the generation time of the bacteria and their mixtures in different proportions. Methods generally used to achieve such parameters are quite complex, tedious and require a lot of time. In Table 2 are summarized the values of k and G of studied bacteria.
From the curve, we can also calculate the amount of heat released (Q) over the culture time:
where A (mW h) is the area, calculated by the trapezoidal method, and K represents a constant whose value is 33.1 J mW−1 h−1, which was calculated from the electric calibration performed by the Joule effect on the equipment.
Also, we have compared the amount of heat exchanged by E. faecalis, P. aeruginosa and their mixtures during the first 24 h (Q 24). In Table 3, the Q 24 by E. faecalis is approximately eight times less than P. aeruginosa, because the metabolic activity of Enterococci is shorter than P. aeruginosa. However, the results showed the mixture 50 % E. faecalis + 50 % P. aeruginosa had a Q 24 lower than expected, due to the interaction between both bacteria.
Discussion
In this paper, we have investigated the interaction relationship between two pathogenic bacteria, E. faecalis and P. aeruginosa, at a concentration of 103 CFU mL−1. We have found that the growth fingerprints of E. faecalis, P. aeruginosa and the mixed microorganisms at different proportions could be real-time monitored using Calvet microcalorimeter equipment. From growth curves of pure and mixed cultures, we have calculated the thermokinetic parameters for evaluating the interaction between two bacteria. When both pathogens were put together, we have observed that in the curves 20 % E. faecalis + 80 % P. aeruginosa and 50 % E. faecalis + 50 % P. aeruginosa, we can detect the presence of P. aeruginosa as bacterial growth is recorded during the duration of the experiment, which is characteristic of this bacterium. However, in both cases, even at low rates of E. faecalis in the sample, the growth profile of P. aeruginosa is modified. Perhaps this is because Enterococcus can use nutrients more effectively or its growth is not completely inhibited by Pseudomonas. In the case of the curve of 80 % E. faecalis + 20 % P. aeruginosa, the growth profiles of both bacteria can be identified more clearly. In the first stretch, we could observe the growth curve of E. faecalis, with three peaks of heat flow, which signal was recorded during the first 10 h of the experiment, and the second stretch where an increase in potential is maintained for the duration of the experiment, which is characteristic of Pseudomonas.
In relation to this work, our group had previously published a study about the mechanism of interaction between E. faecalis and Klebsiella pneumoniae at a concentration of 103 CFU mL−1 [24]. Mixtures of both bacteria were prepared in three different proportions: 20 % E. faecalis + 80 % K. pneumoniae, 50 % E. faecalis + 50 % K. pneumoniae and 80 % E. faecalis + 20 % K. pneumoniae. In this case, unlike the present study, the results revealed that when E. faecalis and K. pneumoniae were mixed, in the curves of samples of 20 % E. faecalis + 80 % K. pneumoniae and 50 % E. faecalis + 50 % K. pneumoniae, the characteristic growth of E. faecalis was not present, due to K. pneumoniae completely inhibiting its growth. While in the sample 80 % E. faecalis + 20 % K. pneumoniae, we can see in the first part of the curve, the characteristic growth of E. faecalis, this is because 80 % of the sample is Enterococci, in the same result as seen in the sample of 80 % E. faecalis + 20 % P. aeruginosa.
Conclusions
Microcalorimetric analysis of heat flow–time curves allows us to investigate the growth of bacteria species as such E. faecalis and P. aeruginosa and their mixtures at low concentrations (103 CFU mL−1). In our study, we have seen that although E. faecalis affects the growth of P. aeruginosa even at low proportions in the sample (20 % E. faecalis + 80 % P. aeruginosa). This could be due to E. faecalis using nutrients from the culture medium in a more effective way than P. aeruginosa, and thus affecting the growth profile. Therefore, microcalorimetry provides a new method to assess the relationship between different bacteria.
References
Braissant O, Wirz D, Göpfert B, Daniels AU. Use of isothermal microcalorimetry to monitor microbial activities. FEMS Microbiol Lett. 2010;303:1–8.
Calvet E, Prat H. Récents Progrès in Microcalorimétrie. Paris: Éditorial Dunod; 1958.
Lago N, Legido JL, Paz-Andrade MI, Arias I, Casás LM. Microcalorimetric study of the growth and metabolism of Pseudomonas aeruginosa. J Therm Anal Calorim. 2011;105:651–5.
Braissant O, Wirz D, Göpfert B, Daniels AU. Biomedical use of isothermal microcalorimeters. Sensors. 2010;10:9369–83.
James AM, editor. Calorimetry past, present and future. In: Thermal and energetic studies of cellular biological systems. Bristol: Wright; 1987. pp. 1–13.
Zaharia DC, Iancu C, Steriade AT, Muntean AA, Balint O, Popa VT, et al. MicroDSC study of Staphylococcus epidermidis growth. BMC Microbiol. 2010;10:322.
Rodriguez D, Daniels AU, Urrusti JL, Wirz D, Braissant O. Evaluation of a low-cost calorimetric approach for rapid detection of tuberculosis and other mycobacteria in culture. J Appl Microbiol. 2011;111(4):1016–24.
Rivero NL, Legido JL, Santos IA, Casás LM. Comparative study of microcalorimetric behavior of Escherichia coli, Proteus mirabilis and Klebsiella pneumoniae. Pol J Microbiol. 2012;61(3):199–204.
Bonkat G, Braissant O, Malte R, Solokhina A, Widmer AF, Frei R, et al. Standardization of isothermal microcalorimetry in urinary tract infection detection by using artificial urine. World J Urol. 2013;31(3):553–7.
O´Neill MAA, Vine GJ, Beezer AE, Bishop AH, Hadgraft J, Labetoulle C, et al. Antimicrobial properties of silver-containing wound dressings: a microcalorimetric study. Int J Pharm. 2003;263(1–2):61–8.
Trampuz A, Salzmann S, Antheaume J, Daniels AU. Microcalorimetry: a novel method for detection of microbial contamination in platelet products. Transfusion. 2007;47:1643–50.
Tan MR, Ren YS, Yan D, Meng XH, Cheng LH, Qiu LL, et al. Detection of microorganisms in different growth states based on microcalorimetry. J Therm Anal Calorim. 2012;109(2):1069–75.
Wang J, Cheng D, Zeng N, Xia H, Fu Y, Yan D, et al. Microcalorimetric study of the effect of Benzoinum and Styrax on the growth of Escherichia coli. Nat Prod Res. 2011;25(4):457–63.
Li XF, Jin C, He J, Zhou J, Wang HT, Dai B, et al. Microcalorimetric investigation of the antibacterial activity of curcumin on Staphylococccus aureus coupled with multivariate analysis. J Therm Anal Calorim. 2012;109(1):395–402.
Luo JY, Yang MH. Demethoxycurcumin: a potential antimicrobial agent. J Therm Anal Calorim. 2014;115(3):2331–8.
Baldoni D, Steinhuber A, Zimmerli W, Trampuz A. In vitro activity of gallium maltolate against Staphyloccocci in logarithmic, stationary, and biofilm growth phases: comparison of conventional and calorimetric susceptibility testing methods. Antimicrob Agents Chemother. 2010;54:157–63.
von Ah U, Wirz D, Daniels AU. Isothermal micro calorimetry—a new method for MIC determinations: results for 12 antibiotics and reference strains of E. coli and S. aureus. BMC Microbiol. 2009;. doi:10.1186/1471-2180-9-106.
Yang LN, Sun LX, Xu F, Zhang J, Zhao JN, Zhao ZB, et al. Inhibitory study of two cephalosporins on E. coli by microcalorimetry. J Therm Anal Calorim. 2010;100(2):589–92.
Entenza JM, Bétrisey B, Manuel O, Giddey M, Sakwinska O, Laurent F, et al. Rapid detection of Staphylococcus aureus strains with reduced susceptibility to vancomycin by isothermal microcalorimetry. J Clin Microbiol. 2014;52(1):180–6.
Braissant O, Chavanne P, de Wild M, Pieles U, Stevanovic S, Schumacher R, et al. Novel microcalorimetric assay for antibacterial activity of implant coatings: the cases of silver-doped hydroxyapatite and calcium hydroxide. J Biomed Mater Res B Appl Biomater. 2014;. doi:10.1002/jbm.b.33294.
Rivero NL, Soto JLL, Santos IA, Casás LM. Differentiation between Staphylococcus aureus and Staphylococcus epidermidis using microcalorimetry. Int J Thermophys. 2013;34:1039–48.
von Ah U, Wirz D, Daniels AU. Rapid differentiation of methicillin-susceptible Staphylococcus aureus from methicillin-resistant S. aureus and MIC determinations by isothermal microcalorimetry. J Clin Microbiol. 2008;46(6):2083–7.
Baldoni D, Hermann H, Frei R, Trampuz A, Steinhuber A. Perfomance of microcalorimetry for early detection of methicillin resistance in clinical isolates of Staphylococcus aureus. J Clin Microbiol. 2009;47(3):774–6.
Vázquez C, Lago N, Legido JL, Arias I, Casás LM, Mato MM. Microcalorimetric study of the growth of Enterococcus faecalis, Klebsiella pneumoniae and their mixtures in an enriched culture medium. J Therm Anal Calorim. 2013;. doi:10.1007/s10973-013-3287-9.
Vázquez C, Lago N, Mato MM, Casás LM, Esarte L, Legido JL, Arias I. Microcalorimetric perfomance of the growth in culture of Escherichia coli, Proteus mirabilis and their mixtures in different proportions. J Therm Anal Calorim. 2014;. doi:10.1007/s10973-013-3535-z.
Engleberg NC, DiRita V, Dermody TS. Schaechter’s mechanisms of microbial disease. 5th ed. Philadelphia: Lippincott Williams & Wilkins; 2013.
Paz Andrade MI. Les Developements Recents de la Microcalorimetrie et de la Thermogenese. 1st ed. Paris: CRNS; 1967.
Verdes PV, Mato MM, Paz Andrade MI, Legido JL. Contribution to study of the thermodynamics properties of mixtures containing 2-methoxy-2-methylpropane, alkanol, alkane. J Chem Therm. 2014;73:224–31.
Lago N, Legido JL, Casás LM, Arias I. Microcalorimetric study of the growth of Enterococcus faecalis in an enriched culture medium. J Therm Anal Calorim. 2012;108:665–70.
Astasov-Frauenhoffer M, Braissant O, Hauser-Gerspach I, Daniels AU, Weiger R, Waltimo T. Isothermal microcalorimetry provides new insights into biofilm variability and dynamics. FEMS Microbiol Lett. 2012;337(1):31–7.
Said J, Walker M, Parsons D, Stapleton P, Beezer AE, Gaisford S. Development of a flow system for studying biofilm formation on medical devices with microcalorimetry. Methods. 2014;. doi:10.1016/j.ymeth.2014.12.002.
Ma J, Qi WT, Yang LN, Yu WT, Xie YB, Wang W, et al. Microcalorimetric study on the growth and metabolism of microencapsulated microbial cell culture. J Microbiol Methods. 2007;68:172–7.
Braissant O, Bonkat G, Wirz D, Bachmann A. Microbial growth and isothermal microcalorimetry: growth models and their application to microcalorimetric data. Thermochim Acta. 2013;555:64–71.
Acknowledgements
We thank María Perfecta Salgado Gonzalez and Sofia Baz Rodríguez for their collaboration with the technical measures. We are also thankful for the financial support provided by the Projects EM 2012/141, CN 2012/285, and “Agrupación Estratégica de Biomedicina (INBIOMED)” by “Xunta de Galicia” and the Project FIS 2011-23322 funded by Ministry of Science and Innovation of Spain. All these projects are co-financed with FEDER funds.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Vazquez, C., Lago, N., Mato, M.M. et al. Microcalorimetric study of the growth of Enterococcus faecalis, Pseudomonas aeruginosa and their mixtures in an enriched culture medium. J Therm Anal Calorim 121, 463–468 (2015). https://doi.org/10.1007/s10973-015-4606-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10973-015-4606-0