
Abstract
The preparation of oligosilsesquioxanes from methyltrimethoxysilane and cyclohexyltrimethoxysilane for use as organic–inorganic fillers is reported. These oligomers were characterized by molecular weight, nuclear magnetic resonance analyses, and Fourier-transform infrared spectroscopy. The poly(methyl methacrylate) nanocomposites containing oligosilsesquioxanes were prepared by two methods: (i) the solution blending method and (ii) the solution–melt blending method. The solution–melt blending method was found to be a superior method for improving the thermal and mechanical properties of PMMA by intermolecular interaction between oligosilsesquioxanes fillers and PMMA.

Highlights
-
Preparation of oligosilsesquioxanes via hydrolysis–condensation reaction of alkylalkoxysilanes.
-
Characterization of oligosilsesquioxanes by nuclear magnetic resonance analyses and Fourier-transform infrared spectroscopy.
-
Difference of poly(methyl methacrylate) composites containing oligosilsesquioxanes by preparation methods.
-
PMMA composite by solution-melt-blending method showed improved properties.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polysilsesquioxanes (RSiO3/2)n are potential raw materials for industrial applications having high thermal, mechanical, and chemical stability caused by the siloxane bonding (Si–O–Si) framework. They are categorized based on their framework as random, ladder, or cage structure [1, 2]. Ladder and cage silsesquioxanes are good target compounds in the field of synthesis and material chemistry because of their highly regulated structures. In general, polysilsesquioxanes are prepared by the sol–gel method using alkoxysilanes. It is an acid- or base-catalyzed hydrolysis–condensation reaction. We previously reported the synthesis of stable poly- and oligosilsesquioxanes [3]. The silsesquioxanes were prepared by the acid-catalyzed hydrolysis–condensation reaction of alkoxysilane under nitrogen flow using a controlled amount of water. The silsesquioxanes were soluble in common organic solvents, such as methanol, acetone, tetrahydrofuran (THF), benzene, and so on, and showed high storage stability over several months. However, our oligosilsesquioxanes were not applied yet as fillers in organic–inorganic composites.
The preparation and application of organic–inorganic hybrid/composite materials are of great interest, as these materials have novel functions and applications that are not common to organics or inorganics [4]. In these studies, organic–inorganic hybrid materials were developed containing various metal oxides, SiO2 being the most important one. Much attention has been paid to modify silica (e.g., methacryloxypropylsilsesquioxane, methylsilsesquioxane, and phenylsilsesquioxane) because of stronger interactions compared with silica nanoparticles [5]. Three methods of producing organic–inorganic hybrid/composite materials containing modified silica have been reported as follows: (i) in situ polymerization [6], (ii) involving chemical bonding between organic segment and modified silica [7], and (iii) physical blending including melt blending [5]. First and second methods are achieved good dispersion of modified silica but complicated. Third method is easy and good cost-effective, compared with the first and second methods. However, this method easily forms macrophase separation. These materials also show better properties than organic polymers containing silica nanoparticles, but have many drawbacks: (i) chemical linking involves synthesis procedures which restrict commercialization, (ii) interparticle interaction often results in phase separation of modified silica particles, and (iii) the influence of visible light absorption of substituents. Therefore, we focus on cyclohexylsilsesquioxane oligomers as inorganic components because we can expect solubility in organic solvents, the easy adjustment of uniform size and shape using the sol–gel method, and strong hydrophobic interactions [8, 9] to stabilize silanols.
In this work, we synthesized oligosilsesquioxanes from methyltrimethoxysilane (MTMS) and cyclohexyltrimethoxysilane (cHTMS) for use as organic–inorganic fillers. The oligosilsesquioxanes were characterized using molecular weights, nuclear magnetic resonance (NMR) analyses, and Fourier-transform infrared (FTIR) spectroscopy. Specifically, the simple hydrolysis–condensation of cHTMS was not reported yet. The nanocomposites composed of poly(methyl methacrylate) (PMMA), which is a widely used polymer, and oligosilsesquioxane sols were prepared by two methods: (i) the solution blending method (SB) [10,11,12,13] and (ii) the solution–melt blending method (SMB) [14].
2 Experimental section
2.1 Measurements
NMR spectra were recorded using a JEOL Resonance JNM-ECP 300 spectrometer (JEOL, Akishima, Japan; 1H at 300.53 MHz and 29Si{1H} at 59.70 MHz). The chemical shifts were reported in ppm relative to chloroform-d (CDCl3) (for 1H: 7.26 ppm in residual chloroform) and tetramethylsilane (for 29Si{1H}: 0.00 ppm) as the internal standards. For the 29Si{1H} NMR spectra, Cr(acac)3 was added to the sample as a paramagnetic relaxation agent. FTIR spectra were recorded on an FT/IR-6100 spectrophotometer (JASCO, Hachioji, Japan), using the KBr disk method, neat, and attenuated total reflectance (JASCO ATR PRO 0450-S, ZnSe prism). Gel permeation chromatography (GPC) was performed on a HPLC system (LC-20AD, Shimadzu, Kyoto, Japan) attached to a PLgel 5 μm Mixed-D column. THF was used as an eluent (1 mL min−1) and RID-20A as the detector at 40 °C. The molecular weight was calculated based on polystyrene standards. Matrix-assisted laser desorption ionization (MALDI) mass spectra were recorded using a reflectron-type time-of-flight mass spectrometer (Bruker, UltraFLEX II), a nitrogen laser (wavelength 337 nm), and trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile as the matrix. Thermal gravimetric analysis (TGA) was performed using a TG-DTA analyzer (2000SE, Netzsch Japan, Yokohama, Japan), applying a heating rate of 10 °C min−1 up to 1000 °C under air atmosphere. The surface hardness was evaluated with the pencil hardness test using a No. 553-M Pencil Scratch Hardness Tester (YASUDA, Nishinomiya, Japan). The test is based on Japanese Industrial Standard JIS K5600-5-4, in which a vertical force of 7.5 N was applied with a pencil to the horizontal film surface at an angle of 45°. The hardness was determined using the hardness values of “Mitsubishi Uni pencils.” Atomic force microscopy (AFM) observations were performed with a SPM-9700 (Shimadzu, Kyoto, Japan).
2.2 Materials
Methanol and THF were purified by a standard process [15] and stored over activated molecular sieves. MTMS and cHTMS were purchased from Tokyo Chemical Industry (Tokyo, Japan) and purified by distillation before use. Hydrochloric acid (6 mol L−1) was purchased from FUJIFILM Wako Pure Chemical Corp. (Osaka, Japan). PMMA (Mw = 999,600 g mol−1; Tg = 117 °C) was purchased from Sigma-Aldrich (Tokyo, Japan) and used as received. The silicon wafer (4″ polishing wafer GlobalWafers Co., Ltd.) was cut to 2.5 cm × 2.5 cm and cleaned three times by ultrasonication in acetone.
2.3 Preparation of oligomethylsilsesquioxane (OMS)
MTMS (13.6 g) and methanol (6.6 g) were placed into a 200 mL four-necked flask equipped with nitrogen inlet and outlet tubes and a mechanical stirrer. The mixture was then stirred at 150 rpm in an ice bath for 10 min. Water and hydrochloric acid were added at the molar ratio of 0.85 for water/silicon and 0.105 for hydrogen chloride/silicon. The mixture was placed in an ice bath and stirred for 10 min, and then again at 22 ± 3 °C for 10 min. This was followed by heating at 70 °C for 3 h under a 360 mL min−1 nitrogen flow. OMS was obtained as a colorless viscous liquid.
2.4 Preparation of oligocyclohexylsilsesquioxane (OcHS)
The cHTMS (2.04 g) and 3.2 g of methanol were placed into a 100 mL four-necked flask equipped with nitrogen inlet and outlet tubes and a mechanical stirrer. The mixture was then stirred at 150 rpm in an ice bath for 10 min. Water and hydrochloric acid were then added at the molar ratios of 1.5–8.0 for water/silicon and 0.105 for hydrogen chloride/silicon. The mixture was placed in an ice bath and stirred for 10 min, and then again at 22 ± 3 °C for 10 min. This was followed by heating at 70 °C for 3 h under a 360 mL min−1 nitrogen flow. OcHS was obtained as a colorless viscous liquid or a white solid.
2.5 Preparation of PMMA–silsesquioxane nanocomposites
The precursor solutions were prepared by mixing PMMA and oligosilsesquioxane sol in THF for 24 h at room temperature (the total weight of PMMA and oligosilsesquioxane was 1.0 g).
For the preparation of thin films, the precursor solution was dropped onto a silicon wafer and spun at the rate of 1000 rpm for 30 s. The thin film was dried at 22 ± 3 °C for 10 min. With regards to the SB method, the thin film was dried at 22 ± 3 °C for 1 day. Regarding the SMB method, the thin film was additionally cured at 150 °C for 1 day.
For the preparation of free-standing films, these precursor solutions were poured into a Teflon box (100 mm × 50 mm × 5 mm), followed by drying at 22 ± 3 °C for 1 day. The free-standing films prepared by the SMB method were additionally cured at 150 °C for 1 day.
3 Results and discussion
3.1 Characterization of OMS
OMS was prepared according to the procedure of our previous report [16, 17]. The results of the hydrolytic polycondensation of MTMS are shown in Table 1.
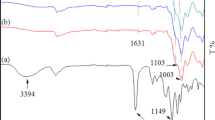
OMS was a viscous liquid. The weight average molecular weight (Mw) was 1800. The signals in the 29Si{1H} NMR spectra appeared in the chemical shift areas ascribed to T1, T2, and T3 unit structures (the symbol Tn(OH)m(OMe)3−n−m denotes the unit structure as RSi(OSi)n(OH)m (OMe)3−n−m (n, m = 1, 2, 3; R = methyl). The 29Si{1H} NMR spectrum of OMS showed four types of silsesquioxane units at −48.1 ppm (T1(OMe)(OH)), −55.9 to −57.0 ppm (T2(OH)), −57.0 to −57.8 ppm (T2(OMe)), and −65.0 to −67.2 ppm (T3) [1, 18, 19], as shown in Fig. 1. From the 1H NMR spectra, the percentage of the remaining methoxy group for OMS was calculated to be 33% (Fig. S1). The FTIR spectrum of OMS is presented in Fig. 2. It indicated the following absorption bands: Si–O–C stretching vibration at 1195 cm−1 [20], branched Si–O–Si stretching vibration at ~1080 cm−1 [21], and linear Si–O–Si stretching vibration at ~1030 cm−1 [22]. It was concluded from the results that OMS formed random-structural oligomers (including liner and cyclic units).

29Si{1H} NMR spectra of a MTMS, b OMS, c cHTMS, d OcHS (Mw = 500), e OcHS (Mw = 800), and f OcHS (Mw = 1600) in CDCl3

FTIR spectra of a OMS by neat method and b OcHS by ATR analysis
3.2 Characterization of OcHS
The results of the hydrolytic polycondensation of cHTMS are summarized in Table 2.
OcHS was a viscous liquid in a molar ratio of H2O/Si = 1.5 and a white solid in molar ratios of 2.5 and 8.0. Increasing the molar ratio of H2O/Si resulted in further hydrolytic polycondensation to produce OcHS with increasing Mw from 500 to 1600. GPC results at various water amounts are shown in Fig. 3. As the water amount increases, the peak shifted towards shorter elution times, indicating the progress of the polycondensation. All the profiles included a sharp single peak in the low molecular weight region, most likely a cyclic compound. In addition, OcHS (Mw 1600) was analyzed by MALDI-TOF-MS as shown in Fig. S2. OcHS (Mw 1600) was a mixture a many silsesquioxane species such as [(C6H11)4Si4O4(OH)4], [(C6H11)5Si5O5(OH)3], [(C6H11)6Si6O7(OH)4], and so on [23, 24]. The signals in the 29Si{1H} NMR spectra appeared in the chemical shift region characteristic of T1, T2, and T3 unit structures (the symbol Tn denotes the unit structure as RSi(OSi)n(OR′)3−n (n = 1, 2, 3; R = cyclohexyl; R′ = Me and H). The 29Si{1H} NMR spectrum of OcHS presented two strong signals assigned to T1 at −48.8 to −54.1 ppm, T2 at −54.4 to −64.0 ppm, and a weaker one for T3 at −66.4 to −73.2 ppm [25], as shown in Fig. 1. Because the hydrolysis reaction improved with increasing H2O/Si ratio, the percentages of the remaining methoxy groups, which were determined from the 1H NMR spectra (Fig. S1), and the area ratio of T1 decreased. The FTIR spectrum of OcHS (Mw 1600) is shown in Fig. 2. Peaks were assigned to cage-like Si–O–Si stretching vibrations at ~1130 cm−1 [26], cyclic Si–O–Si stretching vibrations at ~1060 cm−1 [26], and O–H stretching vibrations at ~3350 cm−1. According to the results, OcHS is a mixture of cyclic and collapsed cage compounds containing hydroxyl group such as [(C6H11)4Si4O4(OH)4]. A possible reaction pathway for the hydrolytic condensation of cHTMS is shown in Scheme 1. This oligosilsesquioxane is used as organic–inorganic filler composited to PMMA.

GPC charts of OcHS with various Mw

A possible reaction pathway for the formation of OcHS
3.3 Characterization and properties of PMMA–oligosilsesquioxane composites
The PMMA–oligosilsesquioxane composites (PMMA–OMS or PMMA–OcHS) were prepared by physical blending methods such as SB and SMB as summarized in Table 3.
The free-standing films of PMMA and oligosilsesquioxane composites are shown in Fig. 4. For visual confirmation, the turbidities of the PMMA–OMS free-standing films prepared according to the SB and SMB methods increased with increasing OMS concentration. In addition, the turbidity of the PMMA–OcHS prepared using the SB method showed a similar trend compared to that of the PMMA–OMS. In contrast, the PMMA–OcHS films prepared by the SMB method showed high transparency. This characteristic suggests the well dispersion and integration of OcHS in PMMA by applying the SMB method.

Photographs of PMMA–oligosilsesquioxane composites a PMMA-10 wt% OcHS by SB method and b PMMA-10 wt% OcHS by SMB method
The free-standing films of PMMA–oligosilsesquioxane composites were characterized by FTIR, the spectra are shown in Fig. 5. The absorption band at 1730 cm−1, assigned to the stretching vibration of the carbonyl group in PMMA, displayed a small shoulder for PMMA–OcHS (SMB method). This was not observed for pure PMMA and PMMA–OcHS prepared by the SB method. The small shoulder was caused by the formation of a hydrogen bond between PMMA and the hydroxyl group of OcHS, as pointed out for a PMMA–SiO2 composite prepared by in situ polymerization [27]. We hypothesized the following: (i) OcHS is dispersed in the PMMA matrix during curing at temperatures higher than the glass transition temperature of PMMA (Tg: 117 °C) and (ii) OcHS in PMMA is immobilized by hydrogen bonds, remaining highly dispersed after curing. In comparison, the νC=O absorption band of PMMA–OMS (prepared by the SB or SMB method) showed no broadening at this region.

FTIR spectra of a pure PMMA, b PMMA–OMS by SMB method, c PMMA–OMS by SB method, d PMMA–OcHS by SMB, and e PMMA–OcHS by SB method using KBr disks
The thermal stability of composites was evaluated by TGA. The results for PMMA and 5% composites are shown in Fig. 6, and the temperatures of 5% weight loss (Td5) are summarized in Table 3. The thermal degradation of pure PMMA with weight loss proceeds in two steps. The first weight loss at 120–250 °C is assigned to the scission of head-to-head linkages [28] or the degradation by radical transfer to the vinyl chain end [29]. The weight loss at 265 °C is caused by radical formation resulting from the cleavage of the main chain, followed by oxidative degradation [30, 31]. The thermal decomposition behavior of PMMA composites prepared by the SB method is similar to that of pure PMMA. On the other hand, the thermal decomposition behavior of PMMA composites prepared by the SMB method differed from that of pure PMMA. The thermal degradation proceeds in one step, with weight loss starting above 240 °C. The scission of head-to-head linkages or vinyl chain ends and the subsequent weight loss does not occur with OcHS and OMS at lower temperature because OcHs and OMS acted as (i) radical scavenger similar to metal oxides [32] and (ii) inhibitor limiting the mobility of the PMMA chain by hydrogen bonding or hydrophobic interactions, respectively.

TGA traces of pure PMMA and PMMA-5 wt% composites
The surface hardness of thin films was evaluated by the pencil hardness test. Surface hardness values are shown in Fig. 7 and listed in Table 3. The pencil hardness is known to be dependent on the adhesion to the substrate, surface smoothness, and degree of chemical/physical crosslinking [33, 34]. Also, the pencil hardness decreases in the presence of phase separation [35]. The surface hardness of PMMA was low because of the rough surface morphology observed by AFM, as shown in Fig. S3. In contrast, PMMA–oligosilsesquioxane thin films revealed an extremely smooth surface. However, the surface hardness was dependent on the content of oligosilsesquioxanes. With regards to PMMA–OMS thin films, 1 wt% OMS provided the highest surface hardness, regardless of blending methods. We proposed that OMS acted as an anchoring layer between PMMA and the substrate. At OMS concentrations higher than 1 wt%, however, the surface hardness decreased and phase separation occurred. The surface hardness of OcHS is weaker than that of OMS, and PMMA-10 wt% OcHS (SMB method) showed the highest surface hardness. This phenomenon suggests that OcHS strongly acted as physical cross-linker because of its high compatibility with PMMA.

Pencil hardness of a PMMA–OcHS by SB method, b PMMA–OcHS by SMB method, c PMMA–OMS by SB, and d PMMA–OMS by SMB method
4 Conclusion
OMS was synthesized by the hydrolysis–condensation of MTMS. The hydrolysis–condensation of cHTMS was investigated by changing the molar ratio of H2O/Si. When H2O/Si was 1.5, OcHS was obtained as a viscous liquid and composed of T1 and T2 units with remaining methoxy groups. When H2O/Si was 2.5 and 8.0, OcHSs were formed as white solids, with few remaining methoxy groups. OcHSs consisted of a mixture of cyclic and collapsed cage compounds. The PMMA nanocomposites with oligosilsesquioxanes as organic–inorganic fillers were prepared by two methods: the SB method and SMB method. Thermal and mechanical analyses showed that the SMB method improved the thermal and mechanical properties of PMMA. Notably, the Td5 and pencil hardness of PMMA-10 wt% was 274 °C and 4 H.
References
Loy DA, Baugher BM, Baugher CR, Schneider DA, Rahimian K (2000) Substituent effects on the sol–gel chemistry of organotrialkoxysilanes. Chem Mater 12:3624–3632
Baney RH, Ito M, Sakakibara A, Suzuki T (1995) Silsesquioxanes. Chem Rev 95:1409–1430
Abe Y, Gunji T (2004) Oligo- and polysiloxanes. Prog Polym Sci 29:149–182
Sanchez C, Philippe B, Michael P, Lionel N (2011) Applications of advanced hybrid organic–inorganic nanomaterials: from laboratory to market. Chem Soc Rev 40:696–753
Zou H, Wu S, Shen J (2008) Polymer/silica nanocomposites: preparation, characterization, properties, and applications. Chem Rev 108:3893–3957
Wang X, Wu L, Li J (2012) Preparation of nano poly(phenylsilsesquioxane) spheres and the influence of nano-PPSQ on the thermal stability of poly(methyl methacrylate). J Therm Anal Calorim 109:323–329
Gunji T, Itagaki S, Kajiwara T, Abe Y, Hatakeyama T, Aoki R (2009) Preparation and properties of siloxane/epoxy organic-inorganic hybrid thin films, self-standing films, and bulk bodies. Polym J 41:541–546
Bizet S, Galy J, Gérard JF (2006) Molecular dynamics simulation of organic–inorganic copolymers based on methacryl-POSS and methyl methacrylate. Polymer 47:8219–8227
Tanaka K, Adachi S, Chujo Y (2009) Structure–property relationship of octa-substituted POSS in thermal and mechanical reinforcements of conventional polymers. J Polym Sci A 47:5690–5697
Tanaka K, Yamane H, Mitamura K, Watase S, Matsukawa K, Chujo Y (2014) Transformation of sulfur to organic–inorganic hybrids employed by networks and their application for the modulation of refractive indices. J Polym Sci A 52:2588–2595
Nguyen QT, Baird DG (2006) Preparation of polymer–clay nanocomposites and their properties. Adv Polym Technol 25:270–285
Hayami R, Wada K, Sagawa T, Tsukada S, Watase S, Gunji T (2017)Preparation and properties of organic–inorganichybrid polymer films using [Ti4(μ3-O)(OiPr)5(μ-OiPr)3(O3PPh)3]·thf. Polym J 49:223–228
Hayami R, Wada K, Nishikawa I, Sagawa T, Yamamoto K, Tsukada S, Gunji T (2017) Preparation and properties of organic–inorganic hybrid materials using titanium phosphonate clusters. Polym J 49:665–669
Motaung TE, Luyt AS, Bondioli F, Messori M, Saladino ML, Spinella A, Nasillo G, Caponetti E (2012) PMMA–titania nanocomposites: properties and thermal degradation behavior. Polym Degrad Stabil 97:1325–1333
Armarego WLF, Chai C (2012) Purification of laboratory chemicals, 7th ed. Butterworth-Heinemann, Oxford, UK
Takamura N, Gunji T, Hatano H, Abe Y (1999) Preparation and properties of polysilsesquioxanes: polysilsesquioxanes and flexible thin films by acid-catalyzed controlled hydrolytic polycondensation of methyl- and vinyltrimethoxysilane. J Polym Sci Part A 37:1017–1026
Gunji T, Kaburagi H, Tsukada S, Abe Y (2015) Preparation, properties, and structure of polysiloxanes by acid-catalyzed controlled hydrolytic co-polycondensation of polymethyl(methoxy)siloxane and polymethoxysiloxane. J Sol-Gel Sci Technol 75:564–573
Hook RJ (1996) A 29Si NMR study of the sol-gel polymerization rates of substituted ethoxysilanes. J Non-Cryst Solids 195:1–15
Sugahara Y, Okada S, Kuroda K, Kato C (1992) 29Si-NMR study of hydrolysis and initial polycondensation processes of organoalkoxysilanes. I. Dimethyldiethoxysilane. J Non-Cryst Solids 139:25–34
Tajima I, Yamamoto M (1987) Characterization of plasma polymers from tetramethylsilane, octamethylcyclotetrasiloxane, and methyltrimethoxysilane. J Polym Sci A 25:1737–1744
Mihelčič M, Surca AK, Kreta A, Gaberšček M (2017) Spectroscopical and electrochemical characterisation of (3-mercaptopropyl)trimethoxysilane-based protective coating on aluminium alloy 2024. Croat Chem Acta 90:169–175
Chen MA, Lu XB, Guo ZH, Huang R (2011) Influence of hydrolysis time on the structure and corrosion protective performance of (3-mercaptopropyl)triethoxysilane film on copper. Corros Sci 56:2793–2802
Pescarmona PP, van der Waal JC, Maschmeyer T (2004) Fast, high-yielding syntheses of silsesquioxanes using acetonitrile as a reactive solvent. Eur J Inorg Chem 2004:978–983
Pescarmona PP, Raimondi ME, Tetteh J, McKay B, Maschmeyer T (2003) Mechanistic study of silsesquoxane synthesis by mass spectrometry and in situ ATR FT-IR spectroscopy. J Phys Chem A 107:8885–8892
Dral AP, Lievens C, ten Elshof JE (2017) Influence of monomer connectivity, network flexibility, and hydrophobicity on the hydrothermal stability of organosilicas. Langmuir 33:5527–5536
Park ES, Ro HW, Nguyen CV, Jaffe RL, Yoon DY (2008) Infrared spectroscopy study of microstructures of poly(silsesquioxane)s. Chem Mater 20:1548–1554
Chan CK, Peng SL, Chu IM, Ni SC (2001) Effects of heat treatment on the properties of poly(methyl methacrylate)/silica hybrid materials prepared by sol–gel process. Polymer 42:4189–4196
Kashiwagi D, Inaba A, Brown JE, Hatada K, Kitayama T, Masuda E (1986) Effects of weak linkages on the thermal and oxidative degradation of poly(methyl methacrylates). Macromolecules 19:2160–2168
Manring LE (1989) Thermal degradation of poly(methyl methacrylate). 2. Vinyl-terminated polymer. Macromolecules 22:2673–2677
Davidson RG (1991) The effect of mechanical degradation on the pyrolysis of poly(methyl methacrylate) and its copolymers. J Anal Appl Pyrolysis 21:181–194
Peterson JD, Vyazovkin S, Wight CA (1999) Stabilizing effect of oxygen on thermal degradation of poly(methyl methacrylate). Macromol Rapid Commun 20:480–483
Tong Y, Lunsford H (1991) Mechanistic and kinetic studies of the reactions of gas-phase methyl radicals with metal oxides. J Am Chem Soc 113:4741–4746
Tadanaga K, Azuta K, Minami T (1997) Preparation of organic–inorganic hybrid coating films from vinyltriethoxysilane–tetraethoxysilane by the sol–gel method. J Ceram Soc Jpn 105:555–558
Takamura N, Okonogi H, Gunji T, Abe Y (2000) Preparation and properties of polysilsesquioxanes—preparation and properties of polymer hybrids from vinyltrimethoxysilane. Kobunshi Ronbunshu 57:198–207.
Hayami R, Wada K, Miyase Y, Sagawa T, Tsukada S, Yamamoto K, Gunji T (2018) Properties and surface morphologies of organic–inorganic hybrid thin films containing titanium phosphonate clusters. Polym J 50:1169–1177
Acknowledgements
This work was supported by JSPS KAKENHI Grant Numbers JP19K05636 and JP18K14287. Dr Kazuhiro Sayama (AIST) is greatly acknowledged for the AFM instrumentation. Prof. Yuichi Negishi and Ms Yukari Imai (Tokyo University of Sci.) are acknowledged for the assistance with MALDI-TOF-MS analysis.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary material
Rights and permissions
About this article

Cite this article
Sato, Y., Hayami, R., Miyase, Y. et al. Preparation and properties of methyl- and cyclohexylsilsesquioxane oligomers as organic–inorganic fillers. J Sol-Gel Sci Technol 95, 474–481 (2020). https://doi.org/10.1007/s10971-020-05291-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-020-05291-2