
Abstract
ZrC–ZrO2 composite ceramic microspheres were prepared by internal gelation combined with carbothermic reduction using fructose as a chelating agent and carbon source. Fructose in the precursor solution formed complex with zirconium ions, which was conducive to the refining of the microstructure of the sintered composite. ZrC–ZrO2 composite with ZrC content as high as 60 wt% could be prepared.

In this paper, fructose was used as a chelating agent and an organic carbon source to prepare ZrCO microspheres by internal gelation and carbothermic reduction. The fructose in the precursor solution could form complex with zirconium ions, which was conducive to the refining of the microstructure of the sintered composite. ZrC–ZrO2 composite with crystal size of ZrO2 and ZrC in nanometer range and ZrC content as high as 60 wt% could be successfully prepared.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Uranium carbon–uranium oxide microspheres have been applied to high-temperature gas-cooled reactors [1, 2] to mitigate the amoeba effect associated with UO2 microspheres [3]. ZrO2 and ZrC can be used as template materials for UO2 and UC. The study of the preparation of ZrC–ZrO2 composite microspheres has important reference value for the preparation of UC–UO2 microspheres [4]. UC–UO2 and ZrC–ZrO2 composite microspheres could be obtained by internal gelation and carbothermic reduction with carbon black as carbon sources [5,6,7,8]. However, the dispersion and particle size of the carbon black had great influence on the microstructure of the carbide microspheres [9], resulting in inhomogeneous distribution of components in the microspheres and potentially decreased crush strength [10].
To refine the microstructure of the microspheres, a water-soluble carbon source, such as sucrose has been tested and ZrC–ZrO2 composite microspheres have been prepared [11]. Such carbon source that is not chemically associated with the metal ions in the sol, however, suffers from the high loss in the washing process of the gel microspheres, resulting in a core–shell structure in the microspheres. Moreover, the increase of sucrose content in the precursor solution also resulted in the decrease of the crush strength. Therefore, it is necessary to explore suitable alternative organic carbon source.
Some references have reported that zirconium ions could form complexes with organic compounds such as acetate [12], acetylacetone [13, 14], and fructose [15, 16]. Tests with a series of organic compounds, including phenolic resin, chitosan, acetyl acetone, glucose, and fructose indicate that fructose was the best material so far for preparation of the composite microspheres. Fructose can be oxidized to gluconic acid in an acid environment, which could form a complex with zirconium ions in solution [16]. In the calcinations process, the complex produces fine particles upon decomposition [17]. So in this paper, fructose was used as a chelating agent and an organic carbon source to prepare ZrC–ZrO2 composite microspheres.
2 Experimental details
2.1 Preparation of ZrC–ZrO2 composite microspheres
The precursor solution with carbon source was prepared according to our previous work [11, 18]. The Zr/Y solution of 28.74 mL including 1.6 mol/L ZrO(NO3)2·xH2O (AR, Aladdin, Shanghai, China) and 0.14 mol/L Y(NO3)3·6H2O (AR, Sinopharm Chemical Reagent Co., Ltd. Beijing, China) was prepared. 2.3 mL concentrated HNO3 (AR, Beijing Chemical Factory, Beijing, China) was added into the Zr/Y solution. Fructose (AR, Aladdin, Shanghai, China) was used as the organic carbon source and was dissolved into the Zr/Y solution under stirring. As the fructose molecule has six carbon atoms, this leads to C/Z molar ratio of 6 when one mole of fructose was added into one mole of ZrO2+. The pH value of the Zr/Y-nitric acid-fructose solutions was less than 1.
Another solution of 20 mL consisting of 3 mol/L hexamethylenetetramine (HMTA) (AR, Sinopharm Chemical Reagent Co., Ltd.) and 2.6 mol/L urea (AR, Sinopharm Chemical Reagent Co., Ltd.) was also prepared.
After cooled to 4 °C for at least 1 h, the above two solutions were mixed to get the precursor solution under magnetic stirring and was further cooled for at least 1 h. The cold precursor solution was dropped into 90 °C silicone oil (AR, Aladdin, Shanghai, China) with a micropipette. Due to the interfacial tension, the liquid droplets spheroidized in the silicone oil and the Eqs. (1) and (2) started. The HMTA is a weak base, which could react with H+ (Eq. 3) generated by the Eq. (2). Then the decomposition of HMTA (Eq. 4) continued to consume H+, initiating the gelation [19]. The liquid droplets were solidified for a few seconds in the silicone oil, and then the gel microspheres were aged for 1 h and allowed to cool overnight.
A washing process was carried out according to Hunt [20] and modified by Gao [11] firstly with trichloroethylene (AR, Aladdin, Shanghai, China) washes to remove the silicone oil on the surface of the gels microspheres, followed by ethanol-thermal treatment at 200 °C for 6 h for reducing the loss of fructose. The microspheres were further washed firstly with NH4OH (AR, Beijing Chemical Factory, Beijing, China) to remove the NH4NO3, urea, and unreacted HMTA from the microspheres; deionized water then was used to remove the NH4OH and other impurities in the microspheres. Finally propylene glycol methyl ether (AR, Aladdin, Shanghai, China) was used to remove the water from the microspheres. After washing, the gel microspheres were allowed to dry for 12 h at 60 °C in an oven.
Heat treatments of the xerogel microspheres were carried out from room temperature to the desired temperatures in the range of 600–1550 °C with a heating rate of 1 °C/min in flowing argon atmosphere for 4 h.
2.2 Characterization
The UV–VIS spectra were recorded for fructose and sucrose solution and their mixture with Zr/Y with a JASCO V-650 spectrometer between 600 and 200 nm. The carbothermic reduction reaction was monitored by thermogravimetry-differential scanning calorimetry (TG-DSC) analysis by NETZSCH STA 449F3 Thermal analyzer, and the test was carried out in argon atmosphere at a heating rate of 10 °C/min from 30 to 1500 °C and with an argon flow rate of 50 mL/min. The X-ray diffraction (XRD) patterns of samples were collected by using a RINT2000 vertical goniometer diffractometer with Cu Kα radiation (Tokyo, Japan). The lattice parameters were refined by Rietveld method from the XRD patterns. The average crystallite size of the samples was calculated by Scherrer’s formula using the XRD patterns. The X-ray photoelectron spectroscopy (XPS) data were collected by PHI Quantro SXM instrument (Ulvac-Phi, Japan). The morphology of the samples was observed on a JSM-6460LV scanning electron microscope (SEM, JEOL, Japan). The sphericity of the microspheres was defined as the ratio of the longest diameter and the shortest diameter of each microsphere, which were measured from the images of the microspheres obtained by a digital optical microscope (ZEISS Stemi 2000-C, Carl Zeiss GmbH, Germany). The average value of one hundred microspheres was taken as the sphericity of the microspheres. The crush strength of the microspheres was measured by KQ-3 crush strength tester. The TC-436 and CS600 (LECO, USA) elemental analysis equipment was used to determine the contents of oxygen and total carbon of the samples.
3 Results and discussion
3.1 The complexing effect of fructose with zirconium ions
UV–Vis studies are commonly used to identify the formation of complexes [12,13,14,15], the UV-Vis absorption spectra of the solutions are recorded and presented in Fig. 1. Water solutions of fructose and sucrose did not show any absorption. Zr/Y-nitric acid solution shows absorption at 298 nm from NO3− [21]. After fructose was added, the absorption blue shifted to 281 nm as a result of oxidation of fructose by nitric acid [16], which formed gluconic acid. A new absorption at 338 nm appeared, which is attributed to the electronic transfer from the zirconium ions to the complexing ligands from gluconic acid [15, 16], as shown in Fig. 2. As a comparison, no absorption existed when sucrose was added to Zr/Y solution, indicating no chemical reactions occurred between zirconium ions and sucrose.

The UV–Vis absorption spectra of the aqueous solutions. a Sucrose solution; b fructose solution; c Zr/Y-nitric acid solution; d Zr/Y-nitric acid-sucrose solution (C/Zr = 6); e Zr/Y-nitric acid-fructose solution(C/Zr = 6)

The complexation of zirconium ions with gluconic acid
3.2 The structural transformation during heat treatment of microspheres
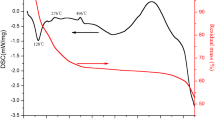
TG-DSC test was conducted to observe the change of the microspheres in heat treatment and to facilitate establishment of the heating program for the dried microspheres. The results are presented in Fig. 3. The TG curve can be categorized into four stages. The weight loss below 200 °C was related to the loss of the H2O, associated with the DSC endotherm near 132 °C, resulting in approximately 15% weight loss. The fast weight loss of the dry spheres between 200 and 600 °C was due to the decomposition of the carbonaceous materials derived from fructose and the residual substances such as HMTA, urea, and NH4NO3. The slower weight loss from 600 to 1300 °C may be related to elimination of structural water, crystallization of c-ZrO2 corresponding to the exothermic peaks were near 734 and 1140 °C in the DSC. The fast weight loss above 1300 °C was due to the carbothermic reduction reaction.

The TG-DSC analysis curves of ZrC–ZrO2 composite dried microspheres in argon atmosphere
Based on the above results, the phase evolution at different heat treatment temperatures was further studied. Figure 4 shows the XRD results of the dried microspheres and those after heat treated at 600–1550 °C in flowing argon with C/Zr being 6 in the precursor solution. The dried microspheres and that calcined at 600 °C were amorphous. The XRD pattern for the ZrC–ZrO2 composite microspheres calcined at 800 °C shows broad and weak diffraction peaks that were due to cubic zirconia (c-ZrO2)(PDF#899069). The only change in the XRD pattern for the ZrC–ZrO2 composite microspheres heat treated at from 800 to 1300 °C was that the c-ZrO2 peaks at 2θ = 30.1°(111), 34.9°(200), 50.2°(220), and 59.6°(311) increased in intensity, indicating the progressive crystallization of ZrO2. The initial ZrC emerged from the sample heat treated at 1400 °C, and the intensity of peaks of c-ZrC phase (PDF#350784) at 2θ = 33.0°(111), 38.3°(200), 55.3°(220), 65.9°(311), and 69.3°(222) increased with temperature. Meanwhile, the intensity of c-ZrO2 gradually decreased with the rise of temperature. The results suggested that ZrC could be produced by carbothermic reduction reaction at and above 1400 °C. ZrC became the main phase at 1550 °C, although a small amount of c-ZrO2 was still present. The carbothermic onset temperature is lower than that of classical preparation of ZrC powder, which occurred at 1657 °C or above [22]. The possible reason for the much lower temperature of formation of ZrC may be that the existence of the complex between the fructose and zirconium ions could achieve homogenous distribution of the precursor materials at molecular level, which dramatically shortens the diffusion path of ZrO2 and C.

X-ray diffraction patterns of the dried ZrC–ZrO2 composite microspheres (C/Zr was 6 in the precursor solution) and microspheres calcined at various temperatures
The average crystallite sizes of ZrC–ZrO2 composite microspheres were calculated by XRD peak analysis based on Scherrer’s equation using the (111), (200), (220), (311), and (222) reflections of ZrO2 and ZrC crystal planes. As shown in Table 1, the average ZrO2 crystallite size increased from 3.20 ± 0.10 to 34.18 ± 3.78 nm with temperature from 800 to 1400 °C. When the heat treatment temperature reached 1400 °C, the crystallite size of c-ZrO2 decreased from 34.18 ± 3.78 to 11.90 ± 3.10 nm with the increase of temperature from 1400 to 1550 °C. Meanwhile, the crystallite size of ZrC in the ZrC–ZrO2 composite microspheres increased from 50.92 ± 1.32 to 56.88 ± 2.88 nm from 1500 to 1550 °C.
3.3 The effect of the fructose content
In order to increase the content of ZrC in the microsphere, the molar ratio C/Zr needs to be adjusted to 3:1 (corresponding to the stoichiometric requirement for carbothermic reduction of zirconia) in the precursor solution. Considering the loss of carbon in the washing process of fructose, the molar ratio of C/Zr was extended to include C/Zr greater than 3, i.e., 1, 2, 3, 4, 5, 6, 7, and 8, respectively.
The XRD patterns of the sintered microspheres calcined at 1550 °C with different amount of fructose are shown in Fig. 5. The results show that the intensity of the ZrC diffraction peaks of the sintered ZrC–ZrO2 composite microspheres increased with increasing content of the fructose, while the intensity of the ZrO2 diffraction peaks gradually decreased. The initial formation of ZrC was observed when C/Zr was 3, and ZrC became the main phase when C/Zr greater than 5, although a weak diffraction peak corresponding toZrO2 was still present.

XRD curves of the ZrC–ZrO2 composite sintered microspheres obtained with various C/Zr molar ratio in precursor solution
The lattice parameters of c-ZrC in the ZrC–ZrO2 composite microspheres with different C/Zr molar ratio in precursor solution was calculated and shown in Fig. 6. The lattice parameters increased with the increase of C/Zr ratio from 3 to 8, approaching that of the pure cubic ZrC phase (a = 4.693 Å, PDF#350784).

The influence of different C/Zr molar ratio on lattice parameter of ZrC in the ZrC–ZrO2 composite microspheres
It is interesting to note that, Fig. 4 shows that the XRD peaks of ZrO2 exhibited a significant shift to the left as the temperature increased when the ZrC phase was observed. Figure 5 also shows that a significant shift of the peaks to the lower angles with the increase of C/Zr ratio. The shifts of the XRD peaks for both phases to lower angles were apparently associated with the carbothermic reaction and the role of carbon in particular. According to the theory proposed in carbothermic reduction of zirconia started with the de-oxygenation of zirconia [23], which releases oxygen and ZrO gas species. Oxygen atom reacts with carbon to form CO, which subsequently reacts with ZrO to form ZrCxOy [23, 24]. ZrCxOy transforms to ZrC with progressive substitution of oxygen by carbon. With the increase of the temperature, the reactions proceed towards the formation of ZrC. When C/Zr is low in the reaction system, substitution of oxygen by carbon could be arrested at an early stage due to the exhaustion of carbon source, leaving relatively more oxygen in the ZrCxOy, which has larger lattice parameter. Only when C/Zr was greater than five in the present system, the amount of carbon could meet the stoichiometric requirement for the near-complete transformation of ZrCxOy to ZrC.
The XPS analysis was conducted to confirm the presence of Zr–C–O in the products. Figure 7a shows the XPS survey spectrum of cross section of ZrC–ZrO2 composite ceramic microspheres with C/Zr = 6 in the precursor solution. The peak of Zr 3d (182 eV), Y 3d(157 eV), O 1s (530 eV), and C 1s (284 eV) confirm the presence of Zr, Y, O, and C atoms in the microsphere. Figure 7b–d displays the XPS narrow scan spectra with fitting curves corresponding to Zr 3d, O 1s, and C 1s bands respectively. Figure 7b shows that the fitting results of Zr 3d band are composed of three components located at 184, 181.5, and 178.4 eV, respectively. The peak at 184 eV is due to the Zr 3d of ZrO2 [25], and the peak at 178.4 eV is attributed to that in ZrC [26]. The peak at 181.5 eV is likely due to the Zr 3d in ZrOxCy [27]. Observation of the spectrum of O 1s (Fig. 7c) clearly demonstrates that there are two components centered at 530.3 and 529 eV corresponding to ZrO2 and ZrOxCy [28], respectively. The spectrum of C 1s (Fig. 7d) is composed of two major peaks at 284.1 and 283.6 eV. The higher binding energy is attributed to elemental carbon [29] and the lower binding energy is due to ZrC [30]. XPS combined with XRD confirms that ZrCxOy is produced in the process of carbothermic reduction.

XPS data for ZrC-ZrO2 composite sintered microsphere with C/Zr = 6 in the precursor solution: a the full survey spectrum; b Zr 3d spectra; c O 1s spectra; d C 1s spectra
The chemical compositions of the sintered ZrC–ZrO2 composite microspheres with different C/Zr ratio were analyzed based on the method in the reference [31]. Figure 8 shows the chemical compositions of the sintered ZrC–ZrO2 composite microspheres with molar ratio of C/Zr in the range of 1–8 in the precursor solution. As shown in Fig. 8 for the ZrC–ZrO2 composite microspheres with C/Zr = 1 and 2, the mass fraction of zirconium carbide was only 9.11 and 12.31wt.%, respectively. Peaks corresponding to ZrC were not observed in the XRD pattern due to the detection error of carbon and oxygen content. For the ZrC–ZrO2 composite microspheres with C/Zr = 3 to 5, the ZrC–ZrO2 composite microspheres had mass fraction of the ZrC of 15.47–36.08wt.%, respectively. Correspondingly, the XRD pattern for the ZrC–ZrO2 composite microspheres with C/Zr 3–5 showed the characteristics peaks of ZrC. When the C/Zr was 6 and above, the amount of ZrC phase increased slowly with the increase of carbon concentration in the precursor solution. The mass fraction of zirconium carbide was greater than 60%.

Variation of the mass fraction of the ZrC of sintered ZrC–ZrO2 composite microsphere with different C/Zr molar ratio
Table 2 presents the crystallite size of the composite microspheres. With the increase of C/Zr in the precursor solution, ZrO2 crystallite size gradually decreased and the crystallite size of ZrC remained almost unchanged at ~60 nm.
3.4 The microstructure of the ZrC–ZrO2 composite microspheres
The surface images of the ZrC–ZrO2 composite sintered microspheres obtained with various C/Zr ratio is displayed in Fig. 9. The images show that all the surfaces of the sintered ZrC–ZrO2 composite microspheres were smooth.

The surface images of the ZrC–ZrO2 composite sintered microspheres obtained with various C/Zr molar ratio in precursor solution: a C/Zr = 1; b C/Zr = 3; c C/Zr = 6; d C/Zr = 8
Figure 10 displays the cross-sectional images of the ZrC–ZrO2 composite sintered microspheres by different C/Zr molar ratio with fructose as carbon source in the precursor solution. As shown in Fig. 10, all the sintered microspheres showed good sphericities of 1.02 ± 0.01. Figure 10 also clearly indicates that when C/Zr = 1, the carbonization of the fructose would form a honeycomb-like skeleton structure, which was consistent with our previous work on the internal structure of the sintered microspheres obtained with sucrose [11]. When C/Zr = 3, the microstructure was obviously refined, with a fine-grained core and slightly coarse-grained shell, which was also observed when sucrose was used. When C/Zr was greater than 4, the microspheres exhibit uniformly fine structure (only the images for C/Zr = 6 and 8 are shown).

The cross-sectional images of the ZrC-ZrO2 composite sintered microspheres obtained with various molar ratio of C/Zr in precursor solution: a, e C/Zr = 1; b, f C/Zr = 3; c, g C/Zr = 6; d, h C/Zr = 8
The formation of core–shell structure when C/Zr ratio was relatively low has been explained as the high solubility of the sucrose and fructose in water [11], which led to loss of carbon in the outer section of the gel microspheres. Comparing the microstructure of the present system with that with sucrose indicates that the loss of carbon source was significantly mitigated and the microstructure of the current system was much refined. The total carbon content of different processing stage of ZrC–ZrO2 composite microspheres (listed in Table 3) confirms this result. There is no obvious change in total carbon content of the as-prepared gel microspheres and that after washing and drying when fructose was used as carbon source, contrasting that a significant loss of carbon occurred after washing when sucrose was the carbon source. Hence, higher carbon was retained in the sintered microspheres, as shown in Table 3.
EDS scan of a microsphere with C/Zr = 6, as shown in Fig. 11, indicates homogeneous distribution of the elements in the microsphere.

Back scattered electron image (a) and the EDS scan (b, c, d) of the cross-section of ZrC–ZrO2 composite sintered microspheres with C/Zr = 6 in the precursor solution
3.5 Effect of fructose on crush strength of ZrCO microspheres
Figure 12 presents the crush strength of the microspheres as a function of C/Zr ratio in the precursor solution. With the increase in C/Zr, the crush strength decreased first and then increased. That the crush strength at C/Zr = 1 was significantly higher than those with C/Zr > 1 was due to the small content of ZrC and the composite was dominantly ZrO2, and the sintering of ZrO2 could be achieved at the heat treatment temperature. With the increase of C/Zr ratio, although ZrO2 was still the main phase in the composite, ZrO2 sintering was inhibited by carbon and isolated ZrC which could not be sintered due to small content in the microsphere. Further increase in C/Zr ratio led to increased ZrC content, and sintering of ZrC could occur, and as a result, a composite structure with ZrO2–ZrC was achieved. The crush strength was still lower than that with C/Zr = 1, suggesting that the sintering of ZrC was not complete [11]. It is natural for such refractory material as ZrC that sintering at 1550 °C would not lead to complete densification, hence the low crush strength. Heat treatment at higher temperature was not carried out due to limitation of our equipment.

Variation of the crush strength of sintered ZrC–ZrO2 composite microsphere with different C/Zr ratio
4 Conclusions
ZrC–ZrO2 composite ceramic microspheres were successfully obtained by combining internal gelation and carbothermal reduction with the fructose acted as chelating agent and carbon source. By using fructose the loss of carbon source was greatly mitigated and the microstructure of the ceramic microspheres was refined. The initial formation of ZrC was apparent after heat treatment at 1400 °C; and ZrC became the main phase after sintering at 1550 °C. The mass fraction of ZrC in the ZrC–ZrO2 composite microspheres could reach more than 60% when the C/Zr ratio was greater than 6 in the precursor solution. XRD patterns and XPS analyses indicated that ZrCxOy was produced in the process of carbothermic reduction, and heat treatment at higher temperatures was required to produce near-stoichiometric ZrC with low oxygen content. The microspheres had homogeneous distribution of ZrC and ZrO2 phases when using the fructose as the carbon source with the C/Zr = 5 and above.
References
Stinton DP, Lackey WJ, Spence RD (1982) Production of spherical UO2-UC2 for nuclear fuel applications using thermochemical principles. J Am Ceram Soc 65(7):321–324
Stinton DP (1983) Mixed uranium dicarbide and uranium dioxide microspheres and process of make same U. S. Patent No. 4,367,184
Barnes CM, Richardson WC, Husser D, Ebner M (2008) Fabrication process and product quality improvements in advanced gas reactor UCO kernels. HTR2008: 58039
Couland M, Fourcaudot S, Abril RJ, Carretero AF, Somer J (2012) Novel production route of yttria-stabilized zirconia fuel kernels and pellets for nuclear fuel applications. J Am Ceram Soc 95(1):133–137
Hunt RD, Lindemer TB, Hu MZ, Del Cul GD, Collins JL (2007) Preparation of spherical, dense uranium fuel kernels with carbon. Radiochim Acta 95(40):225–232
Ma X, Huang X, Kang Z, Zhang G, Luo H (2015) One-pot syntheses and characterization ofzirconium carbide microspheres by carbon microencapsulation Ceram Int 41(5):6740–6746
Phillips JA, Nagley SG, Shaber EL (2012) Fabrication of uranium oxycarbide kernels and compactsfor HTR fuel Nucl Eng Des 251:261–281
Hunt RD, Silva CM, Lindemer TB, Johnson JA, Collins JL (2014) Preparation of UC0.07-0.10N0.90-0.93spheres for TRISO coated fuel particles J Nucl Mater 448(1–3):399–403
Hunt RD, Collins JL (2014) Recommended dispersing conditions for precursor solutions used in the preparation of uranium microspheres with carbon. Ann Nucl Energy 71:1–5
Silva CM, Lindemer TB, Voit SR, Hunt RD, Besmann TM, Terrani KA, Snead LL (2014) Characteristics of uranium carbonitride microparticles synthesized using different reaction conditions. J Nucl Mater 454(1-3):405–412
Gao Y, Ma JT, Zhao XY, Hao SC, Liu B, Deng CS (2016) The preparation of the ZrO2-ZrC ceramic microspheres by the internal gelation process with the sucrose as carbon source. Ceram Int 42(4):4715–4722
Jana S, Biswas PK (1997) Chemical behaviour of zirconium oxychloride octahydrate and acetic acid in precursor solution for zirconia film formation on glass. J Sol–Gel Sci Technol 9:227–237
Georgieva I, Danchova N, Gutzov S, Trendafilova N (2012) DFT modeling, UV–Vis and IR spectroscopic study of acetylacetone-modified zirconia sol–gel materials. J Mol Model 18:2409–2422
Prasanth VG, Prasad G, Kiran T, Rathore RS, Pathak M, Sathiyanarayanan KI (2015) Synthesis, spectral characterization and crystal structure of a new precursor [(CH3COCHCOCH3)2Zr{C6H4(N=CHC6H4O)2}] for nano-zirconia: an investigation on the wettability of polyvinylidene fluoride–nano-zirconia composite material. J Sol–Gel Sci Technol 76:195–203
Li JJ, Jiao XL, Chen DR (2007) Preparation of Y-TZP ceramic fibers by electrolysis-sol-gel method. J Mater Sci 42:5562–5569
Suciu C, Hoffmann AC, Vik A, Goga F (2008) Effect of calcination conditions and precursor proportions on the properties of YSZ nanoparticles obtained by modified sol–gel route. Chem Eng J 138:608–615
Bose S, Wu Y (2005) Synthesis of Al2O3–CeO2 mixed oxide nano-powders. J Am Ceram Soc 88:1999–2002
Gao Y, Ma JT, Zhao XY, Hao SC, Deng CS, Liu B (2015) An improved internal gelation process for preparing ZrO2 ceramic microspheres without cooling the precursor solution. J Am Ceram Soc 98(9):2732–2737
Couland M, Fourcaudot S, JovaniAbril R, Fernandez-Carretero A, Somers J (2012) Novel production route of yttria-stabilized zirconia fuel kernels and pellets for nuclear fuel applications. J Am Ceram Soc 95(1):133–137
Hunt RD, Montgomerya FC, Collins JL (2010) Treatment techniques to prevent cracking of amorphous microspheres made by the internal gelation process. J Nucl Mater 405(2):160–164
Chlistunoff J, Ziegler KJ, Lasdon L, Johnston KP (1999) Nitric/Nitrous acid equilibria in supercritical water. J Phys Chem A 103:1678–1688
Sondhi A, Morandi C, Reidy RF, Scharf TW (2013) Theoretical and experimental investigations on the mechanism of carbothermal reduction of zirconia. Ceram Int 39:4489–4497
David J, Trolliard G, Gendre M, Maître A (2013) TEM study of the reaction mechanisms involved in the carbothermal reduction of zirconia. J Eur Ceram Soc 33:165–179
Gendre M, Maître A, Trolliard G (2011) Synthesis of zirconium oxycarbide (ZrCxOy) powders: Influence of stoichiometry on densification kinetics during spark plasma sintering and on mechanical properties. J Eur Ceram Soc 31(13):2377–2385
Morant C, Sanz JM, Galán L, Soriano L, Rueda F (1989) An XPS study of the interaction of oxygen with zirconium. Surf Sci 218:331–345
Matsuoka M, Isotani S, Sucasaire W, Kuratani N, Ogata K (2008) X-ray photoelectron spectroscopy analysis of zirconium nitride-like films prepared on Si (100) substrates by ion beam assisted deposition. Surf Coat Technol 202:3129–3135
Chu AM, Qin ML, Rafi-ud-din, Zhang L, Lu HF, Jia BR, Qu XH (2013) Carbothermal synthesis of ZrC powders using a combustion synthesis precursor. Int J Refract Met Hard Mater 36:204–210
Steiner SA, Baumann TF, Bayer BC (2009) Nanoscale zirconia as a nonmetallic catalyst for graphitization of carbon and growth of single and multiwall carbon nanotubes. J Am Chem Soc 131:12144–12154
Won YS, Kim YS, Varanasi VG, Kryliouka O, Andersona TJ, Sirimanne CT, McElwee-White L (2007) Growth of ZrC thin films by aerosol-assisted MOCVD. J Cryst Growth 304:324–332
Zhao HS, Liu B, Zhang KH, Tang CH (2012) Microstructure analysis of zirconium carbide layer on pyrocarbon-coated particles prepared by zirconium chloride vapor method. Nucl Eng Des 251:443–448
Gao Y, Ma JT, Zhao XY, Hao SC, Liu B, Deng CS (2016) The fabrication of ZrO2-ZrC microspheres by internal gelation and carbothermal reduction process. J Am Ceram Soc 99(4):1184–1191
Acknowledgements
The authors acknowledge the financial support from Key Program for International S&T Cooperation Projects of China (no. 2016YFE0100700) and National Natural Science Foundation of China (no. 51420105006). “The Thirteenth Five-Year Plan” Discipline Construction Foundation of Tsinghua University (no. 2017HYYXKJS1) is also gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Highlights
-
The fructose was used as a chelating agent and an organic carbon source to prepare ZrC–ZrO2 composite microspheres by internal gelation and carbothermic reduction.
-
The fructose in the precursor solution could form complex with zirconium ions, which was conducive to the refining of the microstructure of the sintered composite.
-
ZrC–ZrO2 composite microspheres with crystallite size of ZrO2 and ZrC less than 100 nm and ZrC content as high as 60 wt% could be successfully prepared.
Rights and permissions
About this article

Cite this article
Sun, X., Ma, J., Chen, X. et al. Sol–gel preparation of ZrC–ZrO2 composite microspheres using fructose as a carbon source. J Sol-Gel Sci Technol 86, 431–440 (2018). https://doi.org/10.1007/s10971-018-4653-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-018-4653-7