
Abstract
Polycrystalline yttria stabilized tetragonal Zirconia (T-ZrO2) fibers were obtained by pyrolysis of gel fibers using zirconium oxychloride octahydrate as raw material. The spinnable zirconia sol was prepared by electrolyzing the zirconium oxychloride octahydrate solution in the presence of acetic acid and sugar (sucrose, glucose or fructose), in which the molar ratio of CH3COOH/ZrOCl2 · 8H2O and sugar/ZrOCl2 · 8H2O was in the range of 1.0–4.0 and 0.2–0.4, respectively. The relation of spinnability to the shape of colloidal particle was discussed. The as-prepared zirconia fibers sintered at different temperatures show smooth and crack-free surface with the diameter of 5–10 μm. Slow heating rate below 600 °C and then sintering at 1,400 °C for 30 min were necessary to obtain the dense tetragonal zirconia ceramic fibers, the particles composed the fibers had the size of ∼150 nm.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
As a widely used ceramics fiber material, the continuous Zirconia (ZrO2) fiber has attracted great interest for several decades as high strength reinforcement material with melting point of over 2,700 °C and excellent chemical resistance to reactive environments, etc [1]. In past decades, many routes to the ZrO2 fibers have been developed. For example, cellulose fibers impregnated with zirconium salts were sintered to remove the organic matrix to form the zirconia fibers [2], and zirconium alkoxide/salts hydrolyzed to form polyzirconoxane [3] or organozirconium complexes [4], which were converted to zirconia fibers. The sol-gel process containing the spinning of the sol from zirconium acetate [5], alkoxide [6, 7] or oxychloride [8, 9] to form the gel fibers and the sintering of gel fibers was a potential method.
Although the sol-gel method was extensively studied, many problems still remained. The greatest limitation to the synthesis of ceramics fibers by the sol-gel process was the expensive precursor, especially the alkoxide compound. The aqueous sol-gel technology was suitable for industrial production, but it was limited in preparing ceramics fibers. The electrolysis of inorganic zirconium salts had several advantages over the conventional sol-gel method [10], in which the expensive metal alkoxide or organometallic complexes could be effectively avoided, the hydrolysis and condensation could be controlled by changing the potential and/or current. Our group [11] reported the preparation of ZrO2 fiber by use of the electrolysis combined with sol-gel method from ZrOCl2 · 8H2O solution using H2SO4 as complexant, however, in that process the quantity of H2SO4 must be controlled exactly to avoid coagulation or precipitate producing, and the presence of SO 2−4 might decrease the quality of the ceramic fiber due to its high decomposing temperature. Therefore it was necessary to develop a more facile and controllable synthetic route to ZrO2 fiber with high quality.
In the present work, the crack-free yttria stabilized T-ZrO2 (YSZ) fibers with smooth surface and the diameter of 5–10 μm were prepared by the electrolysis combined with the sol-gel method. The sugar was used to enhance the stability of sol, in which the carboxyl groups coordinated to Zr(IV) yielded the zirconium acetate complex, favoring the formation of long linear polymer species and spinnable sol.
Experimental
Preparation
ZrOCl2 · 8H2O and Y(NO3)3 · 6H2O (the molar ratio of Y2O3 to ZrO2 was 0.025) were dissolved in distilled water, and the sucrose was added to form a transparent solution. The solution was then heated at 40 °C under magnetic stirring for 1 h, after one third of the needed acetic acid added, the solution was electrolyzed at 25 °C for 48–60 h to obtain the sol. To obtain the spinnable sol, the remained acetic acid was added into the sol after the solution was electrolyzed for ca. 10 h. From the experiment, the optimal molar ratios of CH3COOH/ZrOCl2 · 8H2O and sugar/ZrOCl2 · 8H2O are respectively 2.0 and 0.2, and the concentration of ZrOCl2 · 8H2O is 3.1 mol/L. After the electrolysis process completing, the sol was first aged in air to present a thick film at the surface with the solvent vaporizing, and then aged for 5–8 days in a closed system to form the homogenous sol. A continuous green fiber could be drawn at 1.0 m/s speed by immersing a glass rod with a diameter of ca. 5 mm into the sol and pulling it up, and then dried at 70 °C for 24 h and sintered under air atmosphere in four stages: (1) from room temperature to 600 °C at a rate of 0.5 °C/min, (2) holding at 600 °C for 2 h, (3) raising to designed temperature at a rate of 10 °C/min, (4) holding at the temperature for 2 h. Finally the ZrO2 fibers were obtained after cooling to room temperature naturally.
Characterization
X-ray diffraction (XRD) patterns of the prepared samples were recorded on an X-ray diffractometer (Rigaku D/Max 2200PC) with a graphite monochromator and CuKα radiation (λ = 0.15148 nm) at room temperature while the voltage and electric current were held at 28 kV and 20 mA. Thermal gravimetric (TG) analysis (TGA/SDTA, 851e METTLER) at a rate of 50 mL/min in air flow and heating rate of 20 °C/min was employed to measure the weight loss of the xerogel fibers. The morphology and surface microstructure of the fibers were observed using a scanning electron microscopy (SEM, JEOL JXA-840). IR spectra of the fibers were measured on Fourier transform infrared spectrophotometer (Nicolet, 5DX FTIR) using KBr pellet technique after the fibers were ground into powders. The sol viscosity was analyzed on a cone-and-plate rheometer (RS75 (HAAKE)) at room temperature. The absorption spectra of solution and sol from 200 to 1,100 nm were recorded on an UV–Vis spectrophotometer (Lambda 35, Perkin-Elmer). Nitrogen adsorption-desorption data were measured with an a Quantachrome QuadraSorb SI apparatus at liquid nitrogen temperature (T = −196 °C). Isotherms were evaluated with the Barrett–Joyner–Halenda (BJH) theory to give the surface areas and pore distribution. All specimens for characterization are prepared at the optimal preparation condition: CH3COOH/ZrOCl2 · 8H2O is 2.0, sugar/ZrOCl2 · 8H2O is 0.2, [ZrOCl2 · 8H2O] is 3.1 mol/L, and current density is 450 A/m2.
Results and discussion
Preparation of the spinnable sol
In the present experiment, the optimal electrolysis temperature is 25–35 °C and the suitable current density is 150–450 A/m2. Though the reactions carried out during the electrolyzing process are complicated, the main electrode reactions occur as described in previous report [11], in which the Cl2 and H2 gases are released from positive and negative electrodes respectively. With the electrolysis proceeding, the solution gradually turns into the colorless sol. Further electrochemical reaction will cause the condensation of sol particles, and the linear polymers are formed in presence of acetic acid and sugar. With the electrolysis end-point approaching, the sol becomes viscous and its viscosity increases with aging. As shown in Fig. 1, the spinnable sol exhibits as Newtonian or near-Newtonian flow with the viscosity of ∼10 mPa.s after the precursor solution was electrolyzed at 25 °C for 50 h and then aged at room temperature for 5 days. The transparent green fibers as long as 300 cm could be drawn directly from the spinnable sol.

Relation of the shear stress to the shear rate of the spinnable sol
Table 1 shows the effect of CH3COOH on the spinnability of zirconium sol. When the molar ratio CH3COOH to ZrOCl2 · 8H2O was larger than 1, the sol exhibited excellent spinnability, and the optimal ratio was 1.5–2.0. Further increasing the molar ratio to 4, the sol still possessed good spinnability, but the xerogel fibers became brittle due to the rapid vaporization of excessive acetic acid at the green fiber surface. At the same time, those listed in Table 2 shows that the spinnable sol could be obtained while the molar ratio of sugar to ZrOCl2 · 8H2O was 0.2–0.4 with a suitable ZrOCl2 · 8H2O concentration, and the optimal molar ratio of sugar to ZrOCl2 · 8H2O was 0.2. When the ratio was smaller than 0.1 or larger than 0.6, the white precipitate appeared during the electrolyzing process or sol showed no spinnability after electrolysis, respectively.
It was found that the absence of sugar resulted in the rapid gelation or precipitation during the electrolyzing process, and the sugar played an important role in holding sol stable during the hydrolysis and condensation. Chemical evolution of sugar (sucrose, glucose or fructose) was complicated in the present acidic solution, in which the sucrose molecules firstly hydrolyzed to form d-glucose and d-fructose [12], the fructose and glucose could form an enediol, which coordinated to the zirconyl cations. In order to understand the chemical evolution, the UV–Vis spectra were recorded after the solution was heated at 40 °C for 30 and 70 min and electrolyzed for 15 min, 11 and 24 h, respectively. Before analysis, the solutions were diluted until the ZrOCl2 · 8H2O concentration decreased to 0.005 mol/L. As shown in Fig. 2a, the absorptions at ca. 275 and 335 nm might be attributed to the zirconyl–sugar complex as shown in Fig. 3 [13]. The formation of the zirconyl–sugar complex prevented the precipitation or rapid gelation during electrolysis. However, the zirconyl–sugar complex was destructed progressively with the electrolyzing (Fig. 2b). After electrolyzed for ca. 10 h, the absorptions at ca. 275 and 335 nm almost disappeared and the absorption band at ca. 218 nm might be due to the enediol from the decomposition of zirconium–sugar complex or monose acidification. The IR spectrum of the xerogel fibers (Fig. 5a) shows the C=C stretching vibration at 1,671 cm−1, indicating the decomposition of the sugar-zirconium complex between the zirconium and oxygen. Here, the zirconium acetate was the principal zirconium complex species. As a complexing reagent, the CH3COOH molecules could be used to adjust the spinnability of zirconium sol and prolong the gelation time by coordinating to Zr(IV) [14]. As known, the CH3COOH molecules could coordinate to Zr(IV) by several modes including unidenate, bidentate chelating and bidentate bridging modes [15], which could be distinguished from the frequency separation between the asymmetric and symmetric carboxyl stretching vibrations of COO−. Figure 5b shows the asymmetric and symmetric vibrations of acetate at 1,561 and 1,384 cm−1, and no absorptions of free acetic acid (1,720 and 1,295 cm−1), indicating the acetic acid coordinating to Zr(IV). The frequency separation of 177 cm−1 (between 1,561 and 1,384 cm−1) suggests that the carboxyl group coordinated to zirconium by bidentate bridging mode [15]. The coordination of acetate to Zr(IV) is important in slowing the hydrolysis of Zr(IV) and assisting the complex condensation or polymerization to form the linear polymers for the preparation of the spinnable sol.

UV spectra of zirconyl–sugar complexes after reacted at 40 °C for 30 and 70 min (a), or electrolyzed for 15 min., 11 and 24 h, respectively (b)

The proposed reaction mechanism of zirconyl–sugar complex
The particle shape can be deduced from the rheological properties of the sol [16]. If polymers in the solution are linear or chain-like, the reduced viscosity, η sp /C of a polymer solution can be expressed by Huggin’s Equation (1) [17].
where η sp = η rel−1. η rel = η/η 0, η-viscosity of solution; η 0-viscosity of solvent. Here, η sp, η rel, K C and C are the specific viscosity, the relative viscosity, the proportionality constant and the concentration of the polymer, respectively. The definition of limiting viscosity number [η] is
[η] can be determined by extrapolation of the η sp/C versus C relation to zero concentration of the polymer. The reduced viscosity η sp/C of the solution containing spherical particles can be expressed by the Eq. (3) [18].
K C is a constant, ρ is the density of colloidal particles and C is the concentration of the polymer.
The rheological property of the zirconium sol in Fig. 1 indicated that the sol was Newtonian or near-Newtonian fluid. After the spinnable sol diluted to 0.2, 0.3 and 0.4 mol/L (Zr(IV) concentration), the measured η sp/C values were in agreement with Eqs. (1) and (2). The plot of reduced viscosity versus concentration (Fig. 4) indicated that the η sp/C value increased with the concentration, indicating that particles maintained the linear shape [19].

Relation of viscosity of the sol to its concentration
Evolution of gel fibers
Figure 5 shows the IR spectra of the xerogel fibers and those sintered at different temperatures. The assignment of the absorptions in Fig. 4b is listed in Table 3. The sharp peak at 2,336 cm−1 corresponding to CO2 appears, indicating the absorption of CO2 on the fibers. Upon calcined at 200 °C, the IR spectrum shows no obvious change except the decreasing of the absorption at 1,629 cm−1, although there is ca. 20% weight loss before 200 °C from the TG curve (Fig. 6), indicating that the mass loss of ca. 20% below 220 °C is attributed to the evaporation of the absorbed water and a few of free acetic acid. The shoulder at 1,671 cm−1 is attributed to the C=C stretching vibration, indicating that the enediol is produced from the decomposition of zirconium–sugar complex, which disappears after sintered at 390 °C. The frequencies of the sugar derivative (1,403, 1,453 and 1,350 cm−1 of CH2 bending vibration and the coupling vibrations of OCH, CCH, and COH) in gel fibers shift to lower values in comparison to the corresponding vibrations (1,428, 1,456, and 1,360 cm−1) in free sugar, indicating different status of the sugar derivatives upon coordination to zirconium [20, 21]. After sintering at 390 °C, the bands disappear, suggesting that the sugar derivatives are decomposed at that temperature. The sharp bands at 1,565 and 1,384 cm−1 attributed to the asymmetric and symmetric vibrations of the bridge coordinated COO− almost disappear at 600 °C, indicating that zirconium acetate is decomposed, which is in agreement with the thermal analysis. The TG and DTG curves shown in Fig. 6 demonstrates the marked mass loss of 22% below 220 °C and the mass loss of 6.7% from 220 to 600 °C is due to the decomposition of most zirconium acetate and sugar derivatives. The bands at 1,085, 1,028 and 956 cm−1 (Fig. 5b) indicate the presence of double bonded zirconyl group Zr=O in the gel fibers [22–24]. The absorptions at 650 and 469 cm−1 are assigned to the deformation mode of the acetate groups and Zr–O–Zr vibration, respectively, and those of the acetate groups disappear after heating at 390 °C. The intensity of Zr–O–Zr vibration increases with the calcination temperature rising, and the band at 740 cm−1 does not appear, indicating that no monoclinic ZrO2 forms in the final fibers.

IR spectra of sugar (a), gel fibers (b) and sintered fibers at 100 °C (c), 200 °C (d), 390 °C (e), 440 °C (f), 550 °C (g), 600 °C (h), and 800 °C (i)

TG and DTG curves of the xerogel fiber
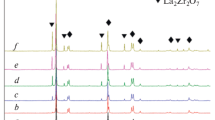
The XRD patterns (Fig. 7) reveal the T-ZrO2 nature of the fibers, which begins to form at 400 °C (JCPDS file: No.48-0224). With the calcined temperature increasing, the peaks change sharper with the increased intensity, showing the increasing of the crystallinity and grain size of T-ZrO2, which is in well agreement with the SEM images.

XRD patterns of the products sintered at different temperatures
Microstructure evolution of the sintered fibers
Figure 8 shows SEM images of the xerogel fibers and those calcined at 900–1,400 °C. The xerogel fibers have the diameter of ca. 15–18 μm, while the diameter of calcined fibers decreases to ca. 5–10 μm due to the shrinkage caused by heat treatment. Both of the xerogel fibers and those calcined ones exhibit uniform diameter without marked defects. The fibers have smooth and dense surface, and the grain size and pores significantly increase with the temperature raising. The individual nano-scale grains are apparent (Fig. 8c) and the grain size significantly increases from 900 to 1,100 °C, but there is a slower grain growth between 1,100 and 1,200 °C. At 1,400 °C, the grains change much coarser and microscopic pores are observed on the fiber’s surface. There is a rapid increasing of the grain size at this temperature, and at this stage, dense ZrO2 ceramics fibers are formed.

SEM images of xerogel fibers (a) and those sintered at different temperatures: 900 °C (b), 1,100 °C (c), 1,200 °C (d), and 1,400 °C (e)
As known, the removing of the volatile substances from the solidified gel fibers at high temperature might produce some defects such as pores, cracks and bubbles, which decrease the fiber strength. In the as-prepared ZrO2 fibers, the cracks can be effectively avoided, and the compact fibers are formed upon calcination. Zarzycki [25] and Scherer [26] have investigated the relationship between the stresses produced during drying and the capillary pores in a gel. They revealed that the larger pores decrease the capillary stresses and make it easier to synthesize the unfractured fibers. To decrease the capillary stresses, the comparative study of the gel fibers drying in air and vacuum is showed in Fig. 9 and Table 4, the green fibers were first dried at 70 °C in air (sample 1) and vacuum (0.1 MPa, sample 2) for 24 h, respectively, then sintered under air atmosphere up to 600 °C at the rate of 0.5 °C/min for 2 h. The pore size distribution of sample 1 and 2 showing that the mean pore diameters are both close to ca. 3.6 nm (Fig. 9), and the surface areas are 52.7 and 41.7 m2/g, respectively. It can be seen that the green fibers drying in air and vacuum conditions at 70 °C have a little difference and slow heating rate before removing the volatile substances is important and efficient for densification. Table 4 obviously shows that the surface area of the fibers significantly decreases with the temperature increasing, and the sample calcined at 1,300 °C shows a high density.

Pore size distribution of zirconia fibers sample 1 and sample 2 (in Table 4), first dried at 70 °C in air and vacuum (0.1 MPa) for 24 h, respectively, then heated in air up to 600 °C at the rate of 0.5 °C/min for 2 h
As densification proceeding, the pores slowly disappear and the drag forces exerted on the grain boundary gradually decrease, and some grains grow fast. So heating the fibers rapidly to a high temperature and holding a short period is indispensable for the densification. Further investigations with more sintering measurement and stricter control grain growth are necessary to optimize the process and improve the properties of the fibers. The experiments show that the grains of sintered ZrO2 fibers at 1,300 °C up to 1,500 °C (Fig. 10) appear the obvious growth. At this stage, the grain size is sensitive to the holding time, especially at 1,500 °C. When the fiber is held for 5 min, the grain size would increase to ∼200 nm, holding for 10 min would result in the formation of ∼350 nm particles and apparent pores. Based on the experiments, the suitable sintering temperature is 1,400 °C and time is 30 min, the average size of the grains in ceramics fibers is ∼150 nm.

Microstructure of sintered zirconia fibers at 1,300 °C for 10 min (a) and 30 min (b), at 1,400 °C for 10 min (c) and 30 min (d), at 1,500 °C for 5 min (e), and 15 min (f ), respectively
Conclusion
The spinnable zirconium sol was prepared by electrolyzing the mixed solution of ZrOCl2 · 8H2O, Y(NO3)3 · 6H2O, sugar and acetic acid. The addition of sugar and CH3COOH increases the sol stability during the electrolysis, the carboxyl group with Zr(IV) complex produces the linear zirconium acetate polymers, which is responsible for the spinnability. Slow heating the gel fibers before 600 °C is necessary to obtain the dense ZrO2 fibers. The obvious grain growth appears from 1,300 to 1,500 °C, and the suitable sintering temperature is 1,400 °C, and the average size of the grains in final fibers is ∼150 nm.
References
Garvie RC, Hannink RH, Pascoe RT (1975) Nature 258:703
Yermolenko IN, Vityaz PA, Ulyanova TM (1985) Sprechsaal 118:323
Abe Y, Kudo T, Tomioka H, Gunji T, Nagao Y, Misono T (1998) J Mater Sci 33:1863
Yogo T (1990) J Mater Sci 25:2394
Marshal DB, Lange FL, Morgan PD (1987) J Am Ceram Soc 70:187
De G, Chatter A, Ganguli D (1990) J Mater Sci Lett 9:845
Emig G., Wirth R (1994) J Mater Sci Lett 29:4559
Blaze JE (1967) US Patent 3322865
Morton MJ, Birchall JD, Cassidy JE (1974) UK Patent 1360199
Manfred P, Klaus A, Klaus-Dieter F, Wolfhang H, Matthias SM (1995) US Patent 5378400
He T, Jiao XL, Chen DR (2001) J Non-Cryst Solids 283:56
Zhang LT (1988) The carbohydrates chemistry. Light industry Press, Beijing, in Chinese
Danielson ND, Heenan CA, Haddadian F (1999) Microchem J 63:405
Aelion A, Loebel A, Eirich F (1950) J Am Chem Soc 72:5702
Nakamoto K (1986) Infrared and Raman spectra of inorganic and coordination compounds. Wiley-Interscience, New York
Sakka S, Yoko T (1992) J Non-Cryst Solids 147&148:394
Huggins ML (1942) J Am Chem Soc 64:2716
Einstein A (1906) Ann Phys 19:289
Sakka S, Kozuka H (1988) J Non-Cryst Solids 100:142
Vasko PD, Blackwell J, Koenig JL (1972) Carbohydrate Res 23:407
Korolevich MV, Zhbankov RG, Sivchik VV (1990) J Mol Struc 220:301
Geiculescu AC, Spencer HG (2000) J Sol–Gel Sci Tech 17(1):25
Komissarova LN, Prozorovskaya ZN, Spitsyn VI (1966) Russ J Inorg Chem 11(9):2035
Straughan BP, Moore W, Mclaughlin R (1986) Spectrochim Acta A 42(4):451
Zarzycki J, Prassas M, Phalippou J (1982) J Mater Sci Lett 17:3371
Scherer GW (1989) J Non-Cryst Solids 109:171
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Li, J., Jiao, X. & Chen, D. Preparation of Y-TZP ceramic fibers by electrolysis-sol-gel method. J Mater Sci 42, 5562–5569 (2007). https://doi.org/10.1007/s10853-006-0979-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10853-006-0979-6