
Abstract
Innovative magnetite–epoxy nanocomposites were prepared starting from magnetite nanoparticles suspended in alcoholic or amino reactive solvents, synthesized by non-hydrolytic sol–gel process from iron (III) acetylacetonate. The obtained suspensions, also synthesized using microwave heating, were mixed with an epoxy monomer (bisphenol A diglycidyl ether, DGEBA), and the formulations were subsequently cured. The thermally activated ring-opening polymerization produced a three-dimensional network in which the suspending medium was covalently linked to the epoxy network according to the chain or step polymerization mechanisms during the cross-linking reaction. This synthetic strategy allowed to obtain nanocomposites in which the nanoparticles play an active role in the polymeric structure, affecting the structural (mechanical and thermal) and functional (magnetic) properties of the final system. The presence of magnetite nanoparticles in the composite resulted in distinct reinforcing effects, acting as rigid filler and/or as cross-linking point, depending on the different chemical environment at the nanoparticle–polymer interphase.
Graphical Abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Polymer-matrix nanocomposites are organic–inorganic hybrid materials in which organic and inorganic components are intimately mixed in the nanometer or submicrometer-dimensional scale [1], allowing to obtain both functional and structural materials in which the mechanical, thermal and processing properties of the polymers are joined to the optical, electrical and magnetic properties of nanofillers.
The inorganic phase provides new functionalities that depend on chemical nature, structure, size and crystallinity of the inorganic nanoparticles, usually metals, metal oxides, nanoclays and nanofibers [2].
The ultimate properties of the composites are dependent on the properties and the volume fraction of components, shape and arrangement of nanoparticles and interfacial interactions between matrix and nanofillers. With the recent development in the nanoscience and nanotechnology fields, the correlation of material properties with filler size has become a focal point of significant interest [3].
Compared to conventional micron-sized particles, nanoparticles have a much higher surface-to-volume ratio. As the particle size decreases, the percentage of molecules/atoms present on the surface is tremendously increased [4]. As a result, particle–particle interactions such as van der Waals and electrostatic forces become stronger, increasing the tendency to aggregation and clusters formation.
With a proper chemical treatment for reducing the surface energy, nanoparticles can be dispersed individually and uniformly distributed in the polymer matrix. In this case, the high surface area leads to enhanced interfacial interactions between the organic and inorganic phases.
For this reason, the structure and properties of the hybrid materials will depend not only on the chemical nature of the components, but also on the bonds type, established at the interphase, strongly influenced by methods and conditions of the synthesis.
The wide choice of the synthetic strategies and the great variety between components and compositions make these systems extremely flexible and able to virtually provide an unlimited number of materials with very different properties and easily adaptable to the experimental requirements [5].
Among the different synthetic strategies, the in situ preparation is a particularly suitable approach because it allows the homogeneous distribution and dispersion of nanoparticles in the polymer matrix, avoiding the problems related to the incorporation of preformed nanofillers by the conventional mechanical mixing. Generally, the in situ approach involves the generation of the nanostructured inorganic oxide directly in a polymer matrix or alternatively the polymerization of an organic monomer in the nanoparticles suspension. The sol–gel chemistry represents one of the preferred ways for the in situ nanoparticle synthesis. The most used method is based on the aqueous (or hydrolytic) sol–gel process but also the so-called non-hydrolytic sol–gel (NHSG) reaction can be used to obtain very pure and crystalline metal oxides [6]. Similarly to the aqueous route, the NHSG process is divided in two steps. The first step (non-hydrolytic reaction) involves the reaction of a metal halide, alkoxide, acetate or acetylacetonate with an organic oxygen donor (such as alcohols, ethers or glycols). The second step (aprotic condensation reaction) can follow different pathways depending on the precursor employed [7]. The flexibility of this synthetic approach is very important because it allows the formation of a chemical environment as much as possible compatible with the organic nature of the polymeric matrix. Indeed, in the NHSG reaction, the solvent not only reacts with the organometallic precursor but also acts as surface ligand, allowing the control of the nanoparticles growth and the functionalization of the nanoparticles surface without the need of any additional reagent [8].
The preparation of polymer-matrix nanocomposites exploiting the NHSG process for the generation of metal oxide nanoparticles was recently proposed in the literature as described in the follows. Poly(methyl methacrylate) (PMMA)/titania composites were recently suggested by Messori et al. exploiting the reaction of TiCl4 and benzyl alcohol or tert-butanol in the presence of PMMA [9, 10]. The synthesis of PMMA/silica composites were also proposed with a very similar approach through the reaction between SiCl4 and ethanol [11]. In comparison with the materials prepared by traditional sol–gel route, these hybrids exhibited higher optical transparency and better thermal stability. Other examples of thermoplastic and thermoset matrix nanocomposites prepared with filler nanoparticles obtained through NHSG process are reported in the literature [12–16].
Recently, an original in situ preparation technique was developed for one step nanocomposites synthesis, directly mixing the resin monomer in the NHSG nanoparticles suspension. The UV light or thermal polymerization produces a three-dimensional structure in which the excess solvent, unreacted in the nanoparticles synthesis, acts both as suspending medium of the nanoparticles and reactive substance toward organic monomers in the subsequent polymerization reaction. This double role of the solvent is the basis of the so-called ‘reactive suspension method,’ used to prepare metal oxide nanoparticles-filled polymers. In [17], titania nanoparticles suspended in benzyl alcohol were mixed with UV-curable cycloaliphatic epoxy resin. Benzyl alcohol was proposed to be covalently linked to the epoxy network, suggesting that the ‘activated monomer’ mechanism [18] was involved during the chain propagation in the cationic ring-opening polymerization. Titanium oxide nanoparticles were also employed to prepare thermally cured epoxy-based nanocomposites, starting from a titania alcoholic suspension [19]. Also in this case, the thermally activated ring-opening reaction developed a polymeric network at which the suspending medium was covalently linked.
In this work, epoxy/magnetite nanocomposites were obtained with the reactive suspension method, starting from the NHSG synthesis of the inorganic nanofillers. Magnetite nanoparticles were prepared from iron (III) acetylacetonate in the presence of alcoholic or amino organic solvent. The so-obtained suspensions were mixed to diglycidyl ether of bisphenol A as is or in the presence of ytterbium (III) trifluoromethanesulfonate as cationic initiator. The alcoholic or amino solvents were then covalently linked to the epoxy network according to the chain- or step-growth mechanism during the propagation step in the ring-opening polymerization. Microscopic, spectroscopic and structural characterization of nanoparticles and microstructural and dynamic thermo-mechanical analysis of nanocomposites were performed with the specific aim to correlate the composite microstructure with functional and structural properties.
2 Experimental
2.1 Materials
Bisphenol A diglycidyl ether (DGEBA, Dow D.E.R.™ 332 with an epoxide equivalent weight of 172–176), benzyl alcohol (BzOH), 2-methyl-hexane-1,3-diol (HD), diethyl triamine (DETA), ytterbium (III) trifluoromethanesulfonate hydrate (Yb(OTf)3) and ethanol (EtOH) were purchased from Sigma-Aldrich (Milan, Italy).
All materials were high purity reactants and were used as received without any further purification.
2.2 Synthesis of magnetite suspensions
1.00 g (2.83 mmol) of iron (III) acetylacetonate (Fe(AcAc)3) prepared as in [20] was dissolved in a 100-mL Schlenk tube with different amount of solvent, in order to evaluate the effect of the precursor-to-solvent ratio on the powder properties. The used solvents were, respectively, BzOH, HD and DETA, whereas the precursor-to-solvent ratio was varied in the molar range 0.01–0.9 (see composition details in Table 1). The reaction was left stirring at room temperature for 15 min and then heated to 200 °C in an oil bath for 48 h.
In the case of microwave-assisted reaction, the synthesis was performed under 2.45 GHz microwave irradiation with a single mode microwave applicator and the temperature was controlled by an optical fiber thermal sensor (Neoptix Reflex four channels, Canada), inserted directly into the reaction mixture, as described in Sciancalepore et al. [21]. All the microwave-assisted reaction parameters (temperature, forward and reflected power) were monitored by means of an Agilent 34970A data acquisition unit.
After reaction, a stable suspension was obtained. To better characterize the inorganic phase, the obtained powders were re-dispersed in methanol (for BzOH and HD) and acetone (for DETA) with an ultrasonic bath and centrifuged at 4000 rpm for 60 min; the powders were washed, centrifuged until the obtainment of a colorless liquid phase and finally dried under reduced pressure.
2.3 Characterization of magnetite powders
Crystalline phases of the synthesized powders were analyzed by X-ray powder diffraction technique using an X’Pert PRO diffractometer (PANalytical, Netherlands), powered by a Philips PW3373/10 Cu LFF DK388689 X-ray generator and fitted with an X’Celerator detector. Diffraction data were acquired by exposing powder samples to Cu–Kα X-ray radiation with a characteristic wavelength of 1.5418 Å. X-rays were generated from a Cu anode supplied with 40 kV and a current of 40 mA. The X-ray diffraction (XRD) patterns were collected at room temperature in 15–75 2θ range, with a step size of 0.0167 and a nominal time per step of 0.008 s−1. Fixed anti-scatter and divergence slits of ½° were used together with a beam mask of 10 mm, and all scans were carried out in ‘continuous’ scanning mode.
Raman scattering experiments were carried out using a micro-Raman system (Labram instrument Jobin Yvon Horiba, Japan) at room temperature. The 632.81 nm line of He–Ne laser with an exposure time of 60 s was used for excitation.
Particles morphology was examined by transmission electron microscopy (TEM, JEM 2010, Jeol, Japan). A drop of the obtained suspensions was placed on a copper grid (200 mesh) covered with PELCO® support films of Formvar, followed by drying. Particles size distribution analysis was performed on the obtained images with the Scanning Probe Imaging Processor (SPIP, Image Metrology A/S, Denmark) software. A minimum of 500 particles were measured for each powder in order to obtain a good statistical approximation. For the determination of the grain size, the equivalent circular diameter ECD (or Heywood diameter) was used. ECD is defined in terms of the projected area A in the image according to ECD = 2(A/π)1/2 and represents the diameter of a circle having an area A equivalent to the nanoparticle shape area.
In order to investigate the presence of residual organic groups on the sample surface, an FT-IR analysis was performed on the obtained powder. The analysis was recorded in the attenuated total reflectance mode with an Avatar 330 spectrometer (Thermo Nicolet, Germany). A minimum of 64 scans with a resolution of 1 cm−1 was implemented.
2.4 Preparation of epoxy–magnetite nanocomposites
Epoxy resins containing Fe3O4 nanoparticles were prepared via chain- or step-polymerization of DGEBA epoxy monomer in the presence of Fe3O4 suspensions synthesized via NHSG process. The formulations were prepared by adding the epoxy monomer to the alcoholic or amine suspension of magnetite nanoparticles. The final Fe3O4 content was in the range between 0.5 and 8 phr (parts of Fe3O4 per hundred parts of resins) (see composition details in Table 2). A typical formulation was prepared by mixing magnetite suspension and epoxy monomer by using a magnetic stirrer (15 min mixing time) and an ultrasonic bath (15 min mixing time). For the alcoholic solvents, Yb(OTf)3 was added as cationic thermal initiator to all the formulations at a concentration equal to 2 phr. The formulations were casted into silicone molds having cavities with dimension 8 × 1 × 0.3 cm3. All the formulations were cured at 100–120 °C for 3 h and post-cured at 130–150 °C for 30 min.
2.5 Characterization of epoxy–magnetite nanocomposites
Dynamic–mechanical thermal analysis (DMTA) was carried out on rectangular strips (average length: 70 mm; average cross section: 20 mm2) by means of a TA Q800 (TA Instruments, New Castle, DE, USA), by employing the dual-cantilever configuration, with a heating rate of 3 °C min−1. The specimens were tested at the frequency of 1 Hz, under a displacement amplitude of 5 μm and a preload of 0.01 N. Storage modulus, E′, and loss factor, tan δ, were measured from −10 °C up to the temperature at which the rubbery state was attained. Glass transition temperature, T g, was assumed at the maximum of the loss factor curves.
T g values, obtained with DMTA analysis were compared with those achieved by the differential scanning calorimetry (DSC). In this case, T g was taken as the mean value of the energy jump of the thermogram (average value between the onset and the endpoint of the glass transition range). DSC analysis was carried out using a Thermal Analysis TA2010 (TA Instruments, New Castle, DE, USA) instrument at a scanning rate of 3 °C min−1 from 10 to 200 °C in N2 atmosphere.
The gel content was determined on the cured composites by measuring the weight loss after 24 h extraction with chloroform at room temperature, according to an adaptation of the standard test method ASTM D 2765-84.
Infrared (FT-IR) analysis was performed on the obtained composites to evaluate the epoxy conversion after the thermal curing. The analysis was performed in the same conditions previously described for the powders.
Transmission electron microscopy (TEM) was carried out on nanocomposite samples in order to evaluate the distribution and dispersion of magnetite nanoparticles into epoxy resin and correlate the structural characteristics with the thermo-mechanical properties of nanocomposites. TEM analysis was performed on a Tecnai 10 electron microscope (FEI Company, USA), using an accelerating voltage of 100 kV. The solid nanocomposite samples were initially trimmed into a shape of trapezoidal flat pyramid with a glass blade and then sectioned into ultrathin slices (100–150 nm) at −30 °C using the cryo-ultra-microtome Leica UC6 (Leica Microsystems, Austria), equipped with a diamond knife (Diatome, USA) working at a knife angle of 35° and with a cutting rate of 300 mm s−1. Sample sections were collected on a 40 % v/v dimethylsulfoxide/water solution and then deposited on 200 mesh copper grids.
3 Results and discussion
3.1 Powder preparation and characterization
The NHSG technique, used for Fe3O4 synthesis, starting from Fe(AcAc)3 as metal oxide precursor, proved to be very versatile and adaptable to the experimental requirements. The effects of reaction time, microwave heating and variation in the precursor-to-solvent ratio were evaluated on the physical and microstructural properties of the obtained nanoparticles. Nanopowders were completely characterized by microscopic, spectroscopic and microstructural techniques. The magnetite phase was detected by X-ray diffractometry and confirmed by Raman spectroscopy. XRD patterns of Mag-BzOH_x powders (chosen as representative) are reported in Fig. 1, which shows that all the synthesized powders are composed by magnetite (JCPDS file 01-075-0449) as crystalline phase, independently on the precursor-to-solvent ratio. In particular, it is quite evident that the peak broadening, commonly observed when the powder has nanometric size, slightly increased as the precursor-to-solvent ratio was increased probably due to the decrease in the crystallite size (Fig. 2). This behavior was also confirmed by the distribution statistics, obtained by the analysis of TEM images. However, the broaden peaks did not allow to completely discard the presence of maghemite, as it possesses similar structures with comparable cell parameters [22]. For this reason, Raman spectroscopy was used allowing to differentiate the iron oxide phases. The Raman spectra of nanoparticles (the spectrum of Mag-BzOH_05 powder shown in Fig. 3 is representative of all samples) exhibit the characteristic bands of magnetite at 668 cm−1 assigned to the A1g transition [23]. TEM analysis (Fig. 4) showed grain sizes ranging from 5 to 20 nm, depending on the precursor-to-solvent ratio and on the used solvent. All the particles size distributions, as extracted by TEM data, have a log-normal behavior, with a more pronounced tail on the larger diameter side and a steep drop on the smaller particle side. Figure 5 compares the size and size distribution of Fe3O4 nanoparticles when the colloidal suspension was prepared with different reactive solvents (molar precursor-to-solvent ratio is fixed). The average grain sizes and the grain size distributions reflect the different reactivity of the considered solvents toward the organometallic precursor. In general, a strongly reactive solvent promotes a high reaction rate and favors the formation of smaller nanoparticles [24]. For example, in the preparation of transition metallic colloids, Reetz et al. [25] found that the size of metallic colloids is strictly dependent on how strong a reduction reagent is, and stronger reducing reagents lead to smaller nanoparticles.

XRD patterns of Mag-BzOH_x powders (the lines are referred to magnetite JCPDS file # 01-075-0449)

Magnification of the (311) XRD peak of the Mag-BzOH_x powders

Raman spectrum of magnetite powders synthesized by NHSG process (sample Mag-BzOH_0.5 as representative)

TEM images of Fe3O4 nanoparticles, obtained using as reactive solvent, respectively, (a) BzOH (Mag-BzOH_1), (b) HD (Mag-HD_1), (c) DETA (Mag-DETA_1) with conventional heating and (d) BzOH in the microwave-assisted synthesis

Size distribution curves obtained with different reactive solvent
DETA can be considered a solvent with a relative high reactivity, and this characteristic results in smaller nanoparticles, with a considerably skewed distribution (Fig. 5). The remarkable asymmetrical distribution occurs because nucleation and growth processes are overlapped in the time scale and, as a consequence, nucleation and growth are not well separated [26] resulting in a nanocrystal size dispersion.
A less reactive solvent induces a low reaction rate and favors relatively larger particles [27, 28]. However, a slow reaction may result in either wider or narrower size distribution. If the slow reaction leads to continuous formation of new nuclei or secondary nuclei, a wide size distribution would be obtained, such as it is observed in the case of HD (Fig. 5).
On the other hand, if no further nucleation or secondary nucleation occurs, a slow reduction reaction would lead to diffusion-limited growth, since the growth of the nuclei would be controlled by the availability of the growth species. Consequently, a narrow size distribution would be obtained. In particular, BzOH allows obtaining relatively small and spherical nanoparticles with a narrow size distribution (Fig. 5).
Within the same solvent, the evolution of the nanoparticles size and size distribution can be observed, when the precursor-to-solvent ratio is changed (Fig. 6 where HD was chosen as solvent). In particular, the nanoparticles average size decreases as the precursor concentration increases (the precursor-to-solvent ratio increases). This behavior is accompanied by increasingly narrow and symmetrical distributions. This behavior was already observed for other experimental synthesis of metallic and alloy nanoparticles. Turkevich, in the synthesis of gold nanoparticles [29], noted an inverse proportionality between the final particles size and the initial concentration of the gold precursor, the chlorauric acid, and demonstrated that a large number of initial nuclei formed in the nucleation stage result in a larger number of nanoparticles with smaller size and narrower size distribution.

Size distribution curves of Fe3O4 nanoparticles, prepared at different precursor concentrations, using HD as reactive solvent
Furthermore, the precursor concentration also influences the nanoparticles morphology. As an example, in the case of nanoparticles synthesized with HD as solvent, at low growth rate, realized at lower precursor concentration (sample Mag-HD_1), nearly round nanoparticles are formed, with broader size distribution (Fig. 7a). At higher growth rate, achieved with a higher precursor concentration (sample Mag-HD_6), a wide variety of anisotropic shapes is obtained, such as trigonal, tetragonal and hexagonal structures (Fig. 7b).

TEM images of Fe3O4 nanoparticles, obtained with HD, as reactive solvent, at different precursor-to-solvent ratio, respectively, Mag-HD_1 (a) and Mag-HD_6 (b)
3.1.1 Influence of microwave heating
The use of microwaves, as heat source, allowed a reduction in the reaction times compared to a traditional heating system [21]. Microwaves led to the reduction in reaction times from days to hours, allowing at the same time a homogeneous heating of the reaction environment [30]. These conditions are ideal for the formation of monodisperse colloids, which require a short and intense nucleation step, followed by time-prolonged and thermally uniform growth [31].
Figure 4d shows, for example, Fe3O4 nanoparticles, obtained with BzOH, using microwaves as heating source; reaction time was 4 h and, if compared with the results obtained after a conventional heat treatment of 48 h (Fig. 4a), the synthetic results are comparable: nanoparticles retain their spherical shape and size under 10 nm.
3.1.2 Influence of the solvent on the surface functionalization of nanoparticles
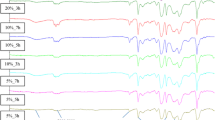
The use of different solvents influenced not only the final size of the nanoparticles, but also the surface functionalization of the Fe3O4 grains. In fact, as showed by FT-IR analysis, nanoparticles obtained with HD as reaction solvent have on their surface in addition to carboxylate groups, arising presumably from the oxidation of a hydroxyl group due to the reduction of the Fe3+ to Fe2+, also free alcohol groups, reasonably present in the same coordinating molecule (Fig. 8b). The bands in the range of 1600–1520 and 1430–1320 cm−1 are assigned, respectively, to the asymmetrical and symmetrical vibrations of the carboxylate group [32], while the broad band at 3300 cm−1 is referred to the free hydroxyl group. In particular, the solvophilic group could act both as compatibilization agent between the Fe3O4 nanocrystals and the organic environment of the resin monomer and as reactive component toward the epoxy moiety of the DGEBA.

FT-IR spectra of the nanoparticles powder, respectively, Mag-BzOH_x (a), Mag-HD_x (b) and Mag-DETA_x (c)
In the case of the amine solvent, the IR spectrum shows no functional group coordinated on the surface of the nanoparticle (Fig. 8c). The absence of surface functionalization will definitely affect the behavior of the nanoparticles in the composite, as shown below.
3.2 Nanocomposite preparation and characterization
In the second step of the nanocomposites preparation, resin polymerization was obtained by means of the in situ approach [33, 34], adding the monomeric precursor and the cationic initiator, if required, directly into the nanoparticle suspension and without any further separation or purification step. When alcohols are used as solvents, the key reaction of the ‘reactive suspension method’ is the so-called ‘activated monomer’ mechanism which can compete with the cationic chain-growth homopolymerization of epoxy resin if OH-containing species are present. According to this reaction, it is possible to covalently incorporate the alcoholic suspending medium into the epoxy-based three-dimensional network.
In the present work, DGEBA was cured using Yb(OTf)3 as a Lewis acid initiator which leads to chain polymerization of epoxy. The reaction mainly proceeds by the cationic ‘active chain end’ (ACE) mechanism depicted in Scheme 1a, in which propagating species are tertiary oxonium ions located at the chain end, where oxirane group of DGEBA is opened by coordination of oxirane oxygen to the initiator and subsequent nucleophilic attack of another oxirane group.

Activated chain end (ACE) and activated monomer (AM) mechanisms
Moreover, the presence of hydroxyl groups can lead to hydroxylic initiated polyetherification processes that can change both the reaction kinetics and the materials properties (Scheme 1b). According to this alternative mechanism, propagation proceeds by nucleophilic attack of oxygen atom in hydroxyl end group on carbon atom in protonated (activated) monomer molecule. This mechanism is known as ‘activated monomer’ (AM) mechanism that should be, in general, less important in extension than the first one, because of the low hydroxyls content.
AM and ACE mechanisms can coexist, being both responsible of polyether macromolecule building. The AM mechanism may outweigh the ACE mechanism when the hydroxyl group concentration is sufficiently high [35]. However, if a monofunctional alcohol, such as BzOH, is present in the reactive suspension during the nanocomposite preparation, the reaction of benzylic hydroxyl groups with oxirane rings determines the interruption of the polymerization along the involved chain by way of the epoxide inactivation (Scheme 2a). This situation results in a less developed three-dimensional network, as revealed by the gel content tests in chloroform, which showed gel fraction values not more than 60 % by weight (Table 3).

Activated monomer mechanism for BzOH (a) and HD (b)
In the case of bifunctional alcohol, such as HD (Scheme 2b), the polymerization is not affected by the presence of the alcohol because the double alcoholic functionality enables the progress of the reaction, leading to the formation of a fully developed three-dimensional network, as shown by the gel content up to 95 % by weight (Table 3).
This result suggests that the AM mechanism occurred during the propagation step in the cationic ring-opening polymerization, allowing the quantitative incorporation of HD within epoxy matrix.
DGEBA monomers can polymerize to form a cross-linked structure also via step-growth polymerization by reacting with amines, phenols, mercaptans, isocyanates or acids. Amines are the most commonly used curing agents/hardeners for epoxides and the case of step-growth polymerizations is mainly represented by epoxy–amine reactions (Scheme 3) that can be considered the key reaction in the ‘reactive suspension method’ for the system Fe3O4–DETA–DGEBA.

Reaction mechanism between epoxide and amine groups
In particular, DETA has five reactive sites and the polymerization reaction between DETA and DGEBA can lead to a highly cross-linked 3D network as shown in Scheme 4.

Reaction mechanism between DETA and DGEBA
3.2.1 Microscopic characterization of magnetite–epoxy resin nanocomposites
TEM analysis was carried out in order to evaluate the distribution and dispersion of magnetite nanoparticles in the polymer matrix (Fig. 9). TEM micrographs of epoxy–magnetite nanocomposites showed the presence of a dispersed phase attributable to the magnetite nanoparticles, as indicated by EDS analysis (here not reported).

TEM micrographs of the magnetite nanocomposites, obtained with different reactive solvents and various precursor-to-solvent ratio. The Fe3O4 content increases from left to right. From up to bottom: Comp-BzOH_1 (a), Comp-BzOH_2 (b) and Comp-BzOH_4 (c); Comp-HD_2 (d), Comp-HD_4 (e) and Comp-HD_6 (f), Comp-DETA_2 (g), Comp-DETA_4 (h) and Comp-DETA_8 (i)
Even if some evident particle agglomeration phenomenon was present, a good enough homogeneous distribution and dispersion of filler were reached for the majority of the samples, avoiding the use of any coupling agent and/or long and complicated dispersion treatments for samples preparation.
It is interesting to note that TEM images obtained with a different magnetite loading exhibited an unexpected trend, taking into account that an increase in the tendency to particle aggregation is usually expected by increasing the filler concentration. On the contrary, in the present case strong aggregation phenomena were clearly evident only in the case of low magnetite content in the composite (Fig. 9a, d, g). Quite surprisingly, a better dispersion state of magnetite nanoparticles in the polymer matrix can be observed for the sample with the higher filler content (Fig. 9c, f, i). This behavior could be ascribed to the formation of a particle surface with a relatively low surface energy and better compatibility with the surrounding organic matrix, which is able to minimize particle–particle interactions. In other terms, the increase in the magnetite content in the nanocomposites is obtained by synthesizing the nanoparticles with an increasingly precursor concentration, keeping constant the amount of reactive solvent. The higher amount of precursor results in the change in the reaction rate and the variation in the nanoparticle growth concerning the dimensions, the shape and the surface structure. At higher reaction rate, nanocrystals have more anisotropic shapes and larger surface areas, which makes them high-energy forms. The nanoparticles are led to decrease their surface energy mainly coordinating and/or absorbing superficially the organic species in the solution. Furthermore, the increased amount of the precursor results, during the synthesis, in the formation of a higher quantity of carboxylate species, bound to the nanoparticle surface, as demonstrated by IR spectra (see Fig. 3 of Supplementary material). These conditions would lead to a self-compatibilization of the system with the formation of a nanoparticle surface having a relatively lower surface energy and better compatibility with the surrounding polymer matrix.
This trend is common to all analyzed systems, regardless of the used reactive solvent. However, in the case of Fe3O4–BzOH–DGEBA and Fe3O4–DETA–DGEBA systems, this behavior is more pronounced because there is the complete dispersion of the nanoparticles in the matrixes with the higher filler loadings (Fig. 9c, i).
In the Fe3O4–HD–DGEBA system, the dispersion tendency of the nanoparticles is lower and small island-like magnetite domains persist even at high magnetite contents (Fig. 9b). In this case, the high surface energy of the nanoparticles is possible to be still the predominant factor in the interface interaction between the nanoparticles and the polymer matrix.
3.2.2 Thermo-mechanical characterization of magnetite–epoxy resin nanocomposites
The influence of the different magnetite load on the mechanical and thermal characteristics of the composites was evaluated by DSC and DMTA. In the nanocomposites obtained with HD as reactive solvent, DSC thermograms showed that the glass transition temperature (T g) was not significantly affected by increasing of the magnetite content (Fig. 10a). The same result was achieved by evaluating the T g, expressed as maximum value of loss factor, with the DMTA analysis (Fig. 10b).

DSC (a) and tan δ (b) curves for Comp-HD_x samples. T g values from DMTA and DSC are also reported as a function of filler content
The fact that T g is not significantly affected by the presence of magnetite in the polymeric network could be due to the interplay between the nanoparticles aggregates, responsible of the reduced interphase volume, and the specific interactions bonded with the organic matrix.
Effective broadening of the tan δ implies a diminishing fraction of free epoxy network chains due to nanoparticles–network chain interaction. The decrement of loss factor tan δ by increasing the magnetite content (Fig. 10b) can be considered as the indirect evidence of the presence of the specific filler–matrix interaction. Kim et al. [36], for example, reported the systematic decrease in the loss factor damping with filler content in the preparation and the characterization of epoxy-based composites containing silica nanoparticles, functionalized with interacting coupling agent.
For the system Comp-HD_x, the storage modulus values, E′, are reported in Fig. 11. The storage moduli measured in the glassy state (that is in the range 0–20 °C) showed only a slight influence due to the presence of the different magnetite amounts. A much stronger E′ variation was observed in the rubbery state. It is very interesting to note that the storage modulus above the glass transition temperature increased significantly by increasing the magnetite content.

Storage modulus values, E′, for the system Fe3O4–HD–DGEBA
The structure and morphology of nanoparticles-filled networks depend on the compatibility of the organic matrix with magnetite nanoparticles, given mainly by the type of their surface functionalization. In fact, magnetite nanoparticles are not completely compatible with components of the organic medium, and show a tendency to aggregation, due to their high surface energy.
As already observed from TEM analysis, for the system Comp-HD_x, the compatibility increases as the nominal concentration of nanoparticles increases, as demonstrated by the lesser tendency to agglomerate. Both the different chemical composition, resulting from the variation in precursor-to-solvent ratio, and the change in the nanoparticles number and size contribute to the evolution of the network structure, affecting thermodynamic factors and kinetic aspects during network formation.
Storage modulus versus temperature curves were used to calculate the reduced storage modulus E′red, the ratio between the moduli of the composites and the matrix in the rubbery region as a function of the filler volume fraction. E′red as a function of magnetite volume fraction was reported in Fig. 12.

Reduced storage modulus dependence on the magnetite particles content (a); in the table experimental and predicted, by generalized Kerner equation, values of reduced storage modulus (b)
To compare the experimental results with data predicted by the Kerner’s model for composite materials [19, 37], the generalized Kerner equation for the reduced modulus of filled polymers was applied in order to evaluate the relative storage modulus dependence on the nanoparticles content. Data reported in Fig. 12 indicate a relevant increment of E′red with respect to the values predicted by the generalized Kerner equation.
Taking into account that in the rubbery region the modulus values are mainly governed by the cross-linking density of the network, this suggests that magnetite nanoparticle aggregates acted not only as rigid reinforcing filler but also as cross-linking points, increasing the cross-linking density of the composite material with respect to the pristine epoxy matrix. Magnetite nanoparticles domains became part of the three-dimensional network, presumably after the cross-linking reaction between the hydroxyl groups present on the nanoparticle surface and the epoxide rings of DGEBA.
Loss factor, tan δ, and storage modulus, E′, for the Comp-DETA_x system are reported in Fig. 13. The thermal–mechanical behavior of this system was completely different respect to the previous one. In fact, in this case the decrease in the glass transition temperature, T g, can be observed, as the Fe3O4 concentration increased (Fig. 13a). The lowering of the T g, associated with the increase in the loss factor, tan δ, is attributable to a decrement in cross-linking density presumably due to a progressive unbalancing between epoxy and amine hardener.

Loss factor, tan δ (a), and storage modulus obtained, E′, (b) and (c) for the Fe3O4–DETA–DGEBA system
Similarly, a systematic decrease in storage modulus, E′, was observed in the rubber region with increasing concentration of magnetite (Fig. 13b).
Moreover, the influence of the increased amount of by-products, which can act as plasticizers, should not be forgotten.
Lastly, magnetite nanoparticles obtained by synthesis between Fe(AcAc)3 and DETA had no surface functionalization, as evidenced by infrared spectroscopy (Fig. 8c) and this condition could determine the lacking interaction of the nanoparticles with the polymer chains at the polymer–nanoparticle interface.
The significant increase in E′, observed in the glassy region up to 4 phr magnetite concentration, indicated the physical reinforcement due to the presence of rigid inorganic nanoparticles (Fig. 13c), dispersed in the epoxy resin individually or as aggregates [38]. Over this concentration, the determining factor in the glassy region on the modulus E′ became the cross-linking degree of the polymeric structure.
The cross-linking density decrease at higher magnetite concentrations affected so deeply the mechanical properties of the composite to be the decisive factor in the composition of the elastic modulus.
4 Conclusions
Suspensions of magnetite nanoparticles in alcoholic or amino reactive solvent, synthesized by means of the NHSG process, were mixed with the DGEBA epoxy monomer, and subsequently cured through a thermally activated polymerization. All organic species (BzOH, HD and DETA) were covalently linked to the epoxy network as reactive co-monomer, suggesting that both chain- and step-growth mechanisms, to produce epoxy polymer, were able to involve other reactive species in the ring-opening polymerization during the reaction propagation.
The nanoparticles size can be varied in the range of 5–20 nm depending on the used solvent and the precursor-to-solvent ratio. The alcoholic solvents BzOH and HD allowed to realize nearly spherical nanoparticles with a narrow and symmetrical size distribution. The nanoparticles obtained with amine solvent DETA had more complex shapes with a broader and asymmetrical distribution.
The presence of magnetite affected significantly both the storage modulus and the loss factor. The dynamic–mechanical and gel content analyses suggested that the changes in the nanocomposites mechanical properties could be ascribed to the presence of nanoparticles, acting as rigid fillers with higher mechanical properties with respect to the epoxy matrix (hydrodynamic effect) and/or as cross-linking densifiers with the increment of cross-linking density of the hybrid organic–inorganic network.
The nanoparticles contribution to the mechanical behavior of nanocomposites could be largely attributed to the different interactions established at the nanoparticle–polymer interface, as a result of the different chemical environment and surface functionalization of nanoparticles.
References
Ajayan PM, Schadler LS, Braun PV (2003) Nanocomposite science and technology. Wiley, New York
Paul DR, Robeson LM (2008) Polymer 49:3187–3204
Jordan J, Jacob KI, Tannenbaum R, Sharaf MA, Jasiuk I (2005) Mat Sci Eng A-Struct 393:1–11
Hiemenz P, Rajagopalan R (2003) Principles of colloid and surface chemistry. Marcel Dekker Inc, New York
Kickelbick G (2007) Hybrid materials: synthesis, characterization and application. Wiley-VCH, New York
Pinna N, Neri G, Antonietti M, Niederberger M (2004) Angew Chem Int Edit 43:4345–4349
Mutin PH, Vioux A (2009) Chem Mater 21:582–596
Pinna N, Antonietti M, Niederberger M (2004) Colloid Surf A 250:211–213
Morselli D, Messori M, Bondioli F (2011) J Mater Sci 46:6609–6617
Morselli D, Bondioli F, Fiorini M, Messori M (2012) J Mater Sci 47:7003–7012
Song X, Wang X, Wang H, Zhong W, Du Q (2008) Mater Chem Phys 109:143–147
Kuan HC, Chiu SL, Chen CH, Kuan CF, Chiang CL (2009) J Appl Polym Sci 113:1959–1965
Wang H, Meng S, Xu P, Zhong W, Du Q (2007) Polym Eng Sci 47:302–307
Jana S, Lim MA, Baek IC, Kim CH, Seok S II (2008) Mater Chem Phys 112:1008–1014
Dirè S, Tagliazucca V, Brusatin G, Bottazzo J, Fortunati I, Signorini R, Dainese T, Andraud C, Trombetta M, Di Vona ML, Licoccia S (2008) J Sol-Gel Sci Technol 48:217–223
Jung K, Bae JY, Park SJ, Yoo S, Bae BS (2011) J Mater Chem 21:1977–1983
Morselli D, Bondioli F, Sangermano M, Messori M (2012) Polymer 53:283–290
Kubisa PJ (2003) J Polym Sci Part A 1(41):457–468
Morselli D, Bondioli F, Sangermano M, Roppolo I, Messori M (2014) J Appl Polym Sci 131:40470–40479
Florini N, Barrera G, Tiberto P, Allia P, Bondioli F (2013) J Am Ceram Soc 96:3169–3175
Sciancalepore C, Rosa R, Barrera G, Tiberto P, Allia P, Bondioli F (2014) Mat Chem Phys 148:117–124
Jubb AM, Allen HC (2010) ACS Appl Mater Interfaces 2:2804–2812
Daou TJ, Pourroy G, Bgin-Colin S, Grenche JM, Ulhaq-Bouillet C, Legar P, Bernhardt P, Leuvrey C, Rogez G (2006) Chem Mater 18:4399–4404
Lieser KH (1969) Angew Chem Int Edit Engl 8:188–202
Reetz MT, Maase M (1999) Adv Mater 11:773–777
Thanh Nguyen TK, Maclean N, Mahiddine S (2014) Chem Rev 114:7610–7630
de Dios M, Barroso F, Tojo C, López-Quintela MA (2009) J Colloid Interface Sci 333:741–748
Zhang Q, Hu Y, Guo S, Goebl J, Yin Y (2010) Nano Lett 10:5037–5042
Turkevic J (1985) Gold Bull 18:86–91
Bilecka I, Niederberger M (2010) Nanoscale 2:1358–1374
Sugimoto T (1987) Adv Colloid Interface 28:65–108
Lu Y, Miller JD (2002) J Colloid Interface Sci 256:41–52
Kalia S, Kang S, Kuma A, Haldora Y, Kumar B, Kumar R (2014) Colloid Polym Sci 9:2025–2052
Mai YW, Yu ZZ (2006) Polymer nanocomposites. Woodhead Publishing Limited, Cambridge
Kubisa P, Penczek S (1999) Prog Polym Sci 24:1409–1437
Kang S, Hong SI, Choe CR, Park M, Rim S, Kim J (2001) Polymer 42:879–887
Kerner EH (1956) Proc Phys Soc B 69:808–813
Yasmin A, Luo JJ, Abot JL, Daniel IM (2006) Comp Sci Technol 66:2415–2422
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Sciancalepore, C., Bondioli, F. & Messori, M. Non-hydrolytic sol–gel synthesis and reactive suspension method: an innovative approach to obtain magnetite–epoxy nanocomposite materials. J Sol-Gel Sci Technol 81, 69–83 (2017). https://doi.org/10.1007/s10971-016-4095-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-016-4095-z