
Abstract
Hybrid organic–inorganic materials exhibit so versatile properties that they can be considered one of the most interesting classes of materials for photonic applications, for the development of both passive and active devices. A synthetic route used for the preparation of nanostructured organic/inorganic (O/I) materials is the assembling of nano-building blocks (NBBs). This approach allows controlling the extent of phase interaction, which in its turn governs the structure-properties relationships. The non-hydrolytic sol–gel process is recognized as a useful route for the preparation of nanostructured molecular units. The condensation reaction of methacryloxypropyl trimethoxysilane and diphenylsilanediol in a non-hydrolytic sol–gel process has been exploited in order to synthesize nanostructured molecular units for the preparation of hybrid organic/inorganic coatings. The non-hydrolytic condensation reactions were run adding different compounds such as triethylamine, titanium isopropoxide, titanium chloride, and dibutyldilauryltin as condensation promoters. The NBB synthesis was also run under controlled hydrolitic conditions, by exploiting the in situ water production using an ethanol/acetic acid mixture. These reactions have been compared in terms of the influence of the employed reagents on the condensation degree and the product structure. Multinuclear NMR, ATR-FTIR and FT-Raman techniques have been used to study the reaction steps and characterize the final condensation products. Hybrid O/I materials have been prepared by assembling methacrylate-based NBBs in the presence of suitable thermal and photo-initiators. The study on the progress of the thermal polymerization process using differential scanning calorimetry (DSC) will be presented, as well as the preliminary results on the two photon polymerization (TPP) process for the preparation of patternable films.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Sol–gel derived hybrid organic/inorganic materials are largely exploited as novel materials, due to the control of the structure at a nanometric scale and often superior properties in respect to polymers. In fact, they guarantee low optical losses, resistance against dry etching processes and scratch, high flexibility in the final material property design, for instance refractive index tunability, and the possibility of embedding large amount of organic chromophores and nanoparticles. Due to their structural flexibility hybrid materials are being increasingly used for a variety of applications, in particular in the field of integrated optics [1–3]. Homogeneously dispersed organic/inorganic hybrid materials can be synthesized by the hydrolytic sol–gel process through hydrolysis-condensation reactions of alkoxide precursors. However, some problems arise from this approach, particularly concerning the appearance of local inhomogeneities in composition and low cross-linking degree in the final material structure. To overcome such drawbacks, in the last two decades, attention has been focused on an alternative route [4]. The exploitation of the non-hydrolytic method is especially useful in the synthesis of hybrid organic/inorganic materials for optical applications, since the residual hydroxyl groups can be reduced or eliminated by forcing the condensation reaction [5]. Together with the advantages of the non-hydrolytic sol–gel process, the selection of suitable precursors allows the preparation of materials with desired properties. In particular, nanosized objects [6] containing lateral organic groups suitable for polymerization can be produced. By this way it is allowed to perform patterning by radiation interaction obtaining high spatial resolution of the created structures [5, 7].
In recent studies we reported on the preparation of hybrid nanostructured units by non-hydrolytic condensation reactions between Ph2Si(OH)2 (diphenylsilanediol, DPDO) and RSi(OEt)3 (trialkoxysilane, R=H, CH=CH2) in presence of pyridine [8, 9]. It was shown that basic condensation promoters were useful to obtain an almost complete reaction between the Si–OH and the Si–OR groups of the precursors (Scheme 1) reaching a good yield in cyclosiloxane-based oligomers.

Condensation reaction between Si–OH and Si–OR groups
Among hybrid materials produced starting from different precursors, those derived from co-condensed organo-alkoxysilanes and organo-silanediol have attracted a lot of attention for electronic and optical applications, particularly in the case of hybrids modified with acrylate functionalities [5, 10–12]. In this work we present a study on the different ability in promoting the non-hydrolytic condensation between methacryloxypropyltrimethoxysilane (MPTMS) and diphenylsilanediol (DPDO), changing the ratio between precursors, solvent and different reagents and catalysts used as condensation promoters. The non-hydrolytic condensation reactions were run in presence of NEt3 (TEA), Ti(OPri)4 (TIPO), titanium chloride, dibutydilauryltin and acetic acid (AA). This acid was used with ethanol as solvent to promote the esterification reaction allowing the in situ production of water. The results of the NBBs spectroscopic characterization by FTIR, Raman and NMR spectroscopies and the comparison with the NBBs prepared by the controlled hydrolytic synthesis, through the in situ generation of water, will be presented. Our research is aimed at the synthesis of NBBs for the production of patternable hybrid films, by exploiting the reactivity of acrylate functionalities in the photopolymerization process. To assess the reactivity of different NBBs towards polymerization, preliminary results on the study of the thermal polymerization by differential scanning calorimetry (DSC) and two photon polymerization (TPP) process will be presented.
2 Experimental
Commercially available reagents (Aldrich, Fluka, ABCR) and absolute ethanol were used without any further purification. Anhydrous tetrahydrofurane was prepared according to the literature [13], and reactions were carried out in N2 atmosphere using standard Schlenk techniques.
2.1 Synthesis procedure
2.1.1 NBBs synthesis
NBB1. Diphenylsilanediol (DPDO, 2.5 mmol, 0.5408 g) was dissolved with THF (50 mmol, 4.06 mL) at RT in a two-neck flask equipped with reflux, and the condensation promoter NEt3 (5 mmol, 0.697 mL) was added to the solution, stirring at 75 °C for 20 min before addition of MPTMS (1.67 mmol, 0.397 mL). Molar ratios between precursors are: DPDO:MPTMS=1.5:1, DPDO:THF:TEA = 1:20:2. The solution was kept stirring at 75 °C for 2 h and aged at RT for 7 days. Then, it was evaporated to dryness at 50 °C under vacuum for 2 h giving a viscous oil, which was dissolved with dry THF to give a final [Si] = 1.85 M.
NBB2. The mixture of DPDO (2.5 mmol, 0.5408 g), THF (50 mmol, 4.06 mL), MPTMS (3.75 mmol, 0.891 mL) and Ti(OPri)4 (0.01472 g, 15.3 μL) was kept stirring for 24 h. Molar ratios DPDO:MPTMS:THF = 1:1.5:20, TIPO = 1 wt.% calculated with respect to total silanes. After aging for 7 days at RT, the clear solution was evaporated to dryness and diluted with THF as reported above.
NBB3. A mixture of DPDO (2.5 mmol, 0.5408 g), MPTMS (12.5 mmol, 2.971 mL), CH3COOH (AA) (5 mmol, 0,286 mL) and ethanol (10 mmol, 4.06 mL) was kept stirring for 14 days at 75 °C. Molar ratio DPDO:MPTMS:AA:EtOH = 1:5:2:4. A viscous yellowish solution was obtained, which was evaporated to dryness and diluted with THF as reported for NBB1.
2.1.2 Hybrid organic/inorganic matrices
Three different types of matrices were prepared. Details on the synthesis are reported elsewhere [14]. For the preparation of the first one, labeled MZrMA, MPTMS (M) was stirred in the presence of acidified water. Zr(OBut)4 (Zr) and methacrylic acid (MA) were separately allowed to react by stirring for 5 min, and the two solutions were mixed together, neutralized with NaOH and kept stirring for one hour, before the addition of the photoinitiator ((E,E,E,E,E,E)-1,13-bis-[4-(diethylamino)phenyl]-tri-deca-1,3,5,6,8,10,12-hexaen-7-one, [15] dissolved in CHCl3, 0.2% with respect to the numbers of acrylic groups) or the addition of the thermal initiator (benzoyl peroxide, BPO, 2 wt.%). The second matrices are based on the use of NBBs. They are prepared in a protocol similar to the first one, but substituting MPTMS with NBB1, NBB2 and NBB3 nanobuilding blocks. The prepared hybrid matrices are labeled NBB1ZrMA, NBB2ZrMA and NBB3ZrMA, respectively. The third type of matrix MZrMAMon was prepared following the procedure described for the first one with the addition of a tri-acrylate monomer labeled “Mon” (tris(2-hydroxy ethyl)isocyanurate-triacrylate, Sartomer® SR368). The ratio between precursors is calculated for different matrices in order to keep constant the number of polymerizable double bonds.
2.2 Characterization techniques
Transmission FTIR spectra were recorded on a Thermo Optics Avatar 550 instrument in transmission mode in the range 4,000–400 cm−1 using KRS5 windows. ATR/FTIR spectra were collected in the range 4,000–550 cm−1, on a Nicolet 870 E.S.P. with a Golden Gate MK2 Diamond Specac cell. Spectra have been recorded positioning the samples on cell platform operating at room temperature (50 scans, 2 cm−1 resolution). 29Si and 13C NMR spectra were recorded with a Bruker Avance 400 spectrometer operating at 79.49 and 100 MHz, respectively, using D2O as external lock and Enrst angle to minimize acquisition time. T1 values were estimated by the null method, according to the literature [16]. In order to avoid the siloxane bonds rearrangements, the THF solutions of the samples (NBB1 and NBB2) were used as such for NMR studies, although the solutions viscosity did not allow to obtain a good spectra resolution. Chemical shifts (ppm) are referenced to tetramethylsilane (TMS), and Si units are labeled according to the usual NMR notation [8, 9]. FT-Raman spectra were recorded on a JASCO, RFT-600 in the 4,000–150 cm−1 range. DSC curves were recorded on a DSC 2010 TA instrument, with a heating rate of 10 °C/min in the range 30–200 °C in N2 flow (100 cc/min).
3 Results and discussion
The ability of diphenylsilanediol to react with trialkoxysilanes leading to oligomeric siloxanes has been demonstrated to depend on the synthesis conditions. Particularly, bases greatly influence the condensation ability through the DPDO deprotonation step, thus favoring the nucleophilic attack to the trifunctional Si alkoxide [8, 9]. The use of pyridine as promoter of the non-hydrolytic condensation reaction between DPDO and MPTMS was unsuccessful, probably due to the lower reactivity of MPTMS compared to vinyltriethoxysilane with the same molar ratio between precursors (silanediol/trialkoxysilane = 1.5/1). The stronger base triethylamine was then used as condensation promoter to obtain sample NBB1. The study of the reaction by FTIR and 29Si NMR spectroscopies showed quite low reaction kinetics with comparison to results previously obtained on vinyltriethoxysilane and triethoxysilane. However, the reaction pathway is the same and proceeds through a first deprotonation step of DPDO by means of the organic base. As previously reported on the reactions between DPDO and vinyltriethoxysilane promoted by pyridine [8], the spectroscopic results show a very low extent of DPDO self-condensation during this first step, with weak signals attributed to the formation of linear and cyclic D units. Accordingly, the water production by DPDO silanol condensation during the reaction with the trialkoxysilane has been considered to give a negligible contribution to the overall condensation process, which is hence considered as a non-hydrolytic process.
We have previously reported on the analysis of the 29Si NMR spectra of self- and cross-condensed species in hybrid systems prepared using modified triethoxysilanes (RTES, R = –H, vinyl) and silanediol [9]. Although it is not trivial to distinguish details in the molecular structure, cyclic species can be identified because their chemical shifts are downfielded with respect to those due to linear compounds because of the reduction of Si–O–Si angle [17].
Classical Dn m and Tn m notations will be used to represent different silicate species, with D and T representing two and three functional silicon, respectively, n the number of oxo bridges and m the number of silicon atoms in the cyclic species.
Figure 1 shows the spectrum obtained mixing DPDO and MPTMS (1.5:1) in THF in the presence of NEt3 (NBB1). In the spectral region characteristic of D-type signals, the resonance at −39.9 is due to terminal D1 units, [9] while the signal at - 47.6 ppm is associated with the linear products, D2 [18] . The signals at −43.5 and −44.1 ppm can be attributed to cyclic D4 units [19, 20]. The ratio between D1 and D2 units was around 1:6, and the D2 products can be separated in linear and cyclic units in a 3:2 ratio (see Table 1). In the T region, correlated resonances due to T2 (−57.7 and −59.7 ppm) and T3 (−62.8, −64.9, and −66.5 ppm) [21, 22] units in a 1:2 ratio are evident. The occurrence of different signals is likely due to the presence of different cages with silicon atoms in different sequences. Table 1 summarizes the chemical shifts and the ratio, estimated from 29Si NMR spectra, between different Dn and Tn units, respectively.

29Si NMR spectra of samples NBB1 (top) and NBB2 (bottom)
Figure 2 shows the 13C NMR spectra of pure MPTMS and NBB1. The main difference between the two spectra is the almost complete disappearance of the Si–OCH3 signal (in the scheme carbon atom 7) and the strong modification and downfield shift of the signal attributed to the methylene carbon of the propyl chain directly linked to silicon (9.4 ppm, –CH2–Si, in the scheme carbon atom 6), as a consequence of the progress in the condensation reaction. In spite of the high TEA/silane ratio used in NBB1 synthesis, no signals attributable to TEA were found in the spectra, proving that the evaporation step under vacuum leads to the complete base removal from the product solution.

13C NMR spectra in THF. From top to bottom: NBB1, NBB2, MPTMS. The scheme reports the labeling of carbon atoms. An asterisk marks the solvent signal
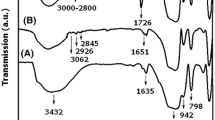
Figure 3a reports the ATR/FTIR spectra of NBB1 obtained by subtracting the THF spectrum to sample spectrum. In the region 1,200–900 cm−1, typical of ν as Si–O–Si stretching bands of condensation products, the spectrum is dominated by a quite broad band characterized by three main components. The main absorption at 1,050 cm−1 is due to linear condensation products. However, the presence of a relatively intense signal at 1,127 cm−1 indicates the formation of cyclic structures, and the absorption at 1,035 cm−1 corresponds to the presence of cyclic T3 products as observed by NMR [20, 23]. In silsesquioxanes resins, the presence of cage type (polyhedral) or network type (cross-linked ladder) structures can be estimated from the position of Si–O–Si signals, due to the siloxane bond variation [24]. The results on NBB1 sample (absorpton at 1,050 cm−1) could indicate a main contribution of network type structures.

ATR/FTIR spectra of samples NBB1 (a) and NBB2 (b)
With the aim to compare the condensation ability of reagents with different chemical nature, metal compounds such as titanium isopropoxide, titanium tetrachloride and dibutyldilauryltin were used. According to spectroscopic results, these compounds, used in catalytic amounts in the syntheses of NBB with DPDO/MPTMS ratio 1.5:1, led to products with very low condensation degree. Better results in network cross-linking were obtained with TIPO, changing the DPDO/MPTMS ratio to 1:1.5 (sample NBB2). The 29Si NMR spectrum (Fig. 1 and Table 1) shows a sligthly higher amount of D2 cyclic units with respect to the sample obtained using TEA (NBB1). However, the 13C NMR spectrum (Fig. 2) presents a residual peak due to Si–OCH3 indicating incomplete condensation, in agreement with the very slow reaction kinetics observed in this case by 29Si NMR and FTIR measurements. The ATR/FTIR spectrum of NBB2 (Fig. 3b) is similar to that of sample prepared with TEA, featuring in the region 1,200–900 cm−1 the broad band due to three main components but with different relative intensities in the two products, probably related to a lower extent of network type structures in NBB2 in comparison with NBB1.
Raman spectra have been collected with the objective to confirm that the prepared NBB are characterized by the presence of siloxane cyclic oligomers. Figure 4 (traces a and b) shows the low frequency range (700–150 cm−1) of the spectrum, where it has been reported that the presence of polysiloxane rings can be observed [25]. The spectra have been normalized with respect to the intensity of the main peak at 1,000 cm−1, which is due to the in plane deformation mode of the phenyl ring of DPDO [26]. The peak at 617 cm−1 is due to the in-plane phenyl ring deformation whereas the peak at 601 cm−1 is attributed to three units cyclosiloxanes. According to the literature [27], the signals below 300 cm−1 could be attributed to network deformation modes of T and D units with the exception of the peak at 377 cm−1 already present in MPTMS spectrum [28]. However, the presence of polysiloxane rings with higher number of silicon units cannot be definitely proved from Raman results. Work is in progress in order to characterize the molecular complexity of NBB1 and NBB2. From preliminary Gel Permeation Chromatography (GPC) measurements, NBB1 is characterized by a broad distribution of products with different molecular weight, from a dimeric species to two main products: a tetramer (Mw 840) and probably a network type structure with Mw 6000. NBB2 GPC curve shows three peaks: a signal at Mw 500 attributable to dimers, the main peak at 1,460 due to an heptamer, which is probably a cage type structure and again a network type structure (Mw 4500).

FT-Raman spectra of NBB1 (a), NBB2 (b) and NBB3 (c) samples
In Fig. 4, the third trace (c) represents the spectrum of the NBB sample NBB3, characterized by a higher load in methacrylate functions (DPDO:MPTMS = 1:5). Indeed, the attempt to synthesize NBBs with higher MPTMS ratio using the same conditions exploited for syntheses NBB1 and NBB2 failed. Therefore, sample NBB3 was prepared taking advantage of the esterification reaction between acetic acid and ethanol, allowing the in situ production of water with very low hydrolysis ratio, thus leading to the evolution of condensation reactions under strictly controlled conditions [29]. Sample NBB3 was obtained as a viscous solution, which can be dried giving a gel that can be redissolved in organic solvents like THF. The FTIR (not shown) and Raman spectra of NBB3 highlighted the formation of siloxane bonds at the expense of both SiOH and SiOCH3 bonds, respectively, from DPDO and MPTMS, and the preservation of the methacrylate groups. The network crosslinking can be evaluated from the 29Si SP-MAS spectrum, which was recorded on the dried gel powders and presents sharp peaks at −38 ppm (D1), −42 ppm (D2 cycles), −48 ppm (D2), −58 ppm (T2) and −66 ppm (T3). The preliminary GPC measurement on NBB3 shows a single main signal centered at Mw 1525, which can be attributed to an heptamer in a cage like structure and small contributions due to dimers. Thus, the GPC results show that the synthesis approach based on the in situ water production is much more effective in controlling the size of the molecular units. The complete characterization of NBB3 will be reported elsewhere.
The reason for obtaining NBBs with higher number of reactive organic functions stays in the use of these nano-objects for the preparation of polymerizable hybrid organic/inorganic materials. The use of highly reactive nano-objects can be a promising way to reach high spatial resolutions in film patterning, and this can be related to the availability of reactive organic functions. The activity of NBBs has been investigated by thermal and two photon induced polymerization (TPP). To this end film samples have been prepared according to a protocol developed in [14], using a BPO, as thermal initiator, and (E,E,E,E,E,E)-1,13-bis-[4-(diethylamino)phenyl]-tri-deca-1,3,5,6,8,10,12-hexaen-7-one, as photoinitiator. The study of the thermal polymerization process has been performed analysing the DSC traces of five different samples. The data of ΔH (J/g) and peak maximum temperature, calculated from DSC curves are reported in Table 2. The DSC curve relative to sample MZrMA is characterized by a very low and broad exothermal effect. Matrices prepared with different NBBs and the triacrylate monomer display, in the temperature range 90–160 °C, a more intense polymerization effect featuring two components, a main peak and a shoulder at high temperature. These two components account for two families of reactive double bonds. The position and intensity of the main peak depend on the composition of matrices. A decrease in peak position and an increase in intensity is observed moving from the MZrMA matrix, the matrices prepared with NBB1, to NBB2, to NBB3, and finally to the matrix prepared with the branched triacrylate monomer, MZrMaMon. This trend indicates a more effective polymerization process in the order indicated above and is dependent on the availability and mobility of the reactive methacrylate functions in the hybrid network.
The TPP has been performed on spin coated films prepared with the matrices listed in Table 2. The photopolymerizationn has been obtained using a Ti:Sapphire femtosecond laser, operating at 850 nm at different incident energies. The laser beam is focused on the sample by using a microscope objective 50× with 0.8 numerical aperture. Different solvents have been used as developers of the photopolymerized structures, after laser irradiation [14]. The availability of a high number of reactive organic groups on nano-objects of controlled sizes seems to play an important role in the preparation of well defined surface figures. In these terms, NBB3ZrMA matrix allows to reach good results with the preparation of well resolved spots, particularly for low incident energies (Fig. 5), whereas matrices prepared with NBB1 and NBB2 nanobuilding blocks led to low cross-linking of the exposed area after laser irradiation. Good results have been obtained also with MZrMaMon matrices (Fig. 5), confirming the importance of the presence of a large double bond numbers.

Confocal images of the spots obtained by TPP after development stage of sample MZrMAMon (left) and NBB3ZrMA, irradiated at 850 nm, at two different incident energies
4 Conclusions
Nanostructured molecular units of different molecular complexity were synthesized starting from methacryloxypropyltrimethoxysilane and diphenylsilanediol using the non-hydrolytic condensation reaction with suitable promoters. With low amount of methacrylate groups, TEA (NBB1) and TIPO (NBB2) were effective in promoting condensation. However, the non-hydrolytic condensation promoted by TEA or TIPO was not successful in achieving NBBs with a high load of reactive organic groups. Moreover, NBB1 and NBB2 appear characterized by a broad distribution of products and the formation of network type structures. On the contrary, a different synthetic approach, based on the in situ water production by addition to the silane precursors of an acetic acid/ethanol mixture, allowed preparing a new NBB with DPDO/MPTMS ratio 1:5, characterized by a better size control of the nano-objects (NBB3).
The obtained samples were used to prepare hybrid O/I matrices by polymerization of the organic functions. A relationship between the structure and the precursor ratio in the pristine NBBs and the polymerization yield was established on the basis of the DSC study on thermal polymerization, concluding that NBB3 allows preparing highly cross-linked networks.
This result is in agreement with the experimental evidence acquired in the study on TPP process. Patternable films have been obtained from matrices prepared with different NBBS. The comparison of the results of the TPP of these matrices indicates NBB3 as the best NBB for the realization of well defined and small structures.
References
Sanchez C, De Soler-Illia AAGJ, Ribot F, Grosso D (2003) C.R. Chimie 6:1131
Houbertz R, Frohlich L, Poall M, Streppel U, Dannberg P, Brauer A, Serbin J, Chichkov BN (2003) Adv Eng Mater 5:551
Ribeiro SJL, Messaddeq Y, Goncalves RR, Ferrari M, Montagna M, Aegerter MA (2000) Appl Phys Lett 32:67
Arkles B, MRS Bulletin, May 2001, 402–407
Buestrich R, Kahlenberg F, Popall M, Dannberg P, Müller-Fiedler R, Rösch D (2001) J Sol–Gel Sci Technol 20:181
Schubert U (2004) J Sol–Gel Sci Technol 31:19
Innocenzi P, Lebeau B (2005) J Mater Chem 15:3821
Dirè S, Egger P, Di Vona ML, Trombetta M, Licoccia S (2004) J Sol–Gel Sci Technol 35:57
Di Vona ML, Trombetta M, Dirè S, Egger P, D’Ottavi C, Licoccia S (2005) J Sol–Gel Sci Technol 35:151
Nam K-H, Lee T-H, Bae B-S, Popall M (2006) J Sol–Gel Sci Technol 39:255
Bae B-S (2004) J Sol–Gel Sci Technol 31:309
Houbertz R, Schulz J, Frönlich L, Domann G, Popall M (2003) Mater Res Soc Symp Proc 769:H7.4.1
Furniss BS, Hannaford AJ, Smith PWG, Tatchell AR (1989) Texbook of practical organic chemistry Vogel’s, V edn. Longman Scientific & Technical
Fortunati I, Dainese T, Signorini R, Bozio R, Tagliazucca V, Dirè S, Lemercier G, Mulatier J-C, Andraud C, Schiavuta P, Bottazzo Y, Della Giustina G,Brusatin G, Guglielmi M (2007) Proc of SPIE 6645:664520
Martineau C, Lemercier G, Andraud C, Wang I, Bouriau M, Baldeck M (2003) Synth Met 138:353
Martin ML, Delpuech JJ, Martin GJ in Practical NMR Spectroscopy Heyden & Son Ltd. London 1980
Devreux F, Boilot JP, Chaput F (1990) Phys Rev A 12:6901
Mayer BX, Zollner P, Kahlig H (1999) J Chromatogr A 848:251
Mayer BX, Zöllner P, Rauter W, Kählig H (2001) J Chromatogr A 917:219
Brown F, Vogt LH (1965) J Am Chem Soc 87:4313
Sellinger A, Laine RM (1996) Chem Mater 8:1592
Sellinger A, Laine RM (1996) Macromolecules 29:2327
Sassi Z, Bureau JC, Bakkali A (2002) Vib Spectrosc 28:251
Doux C, Aw KC, Niewoudt M, Gao W (2006) Microelectron Eng 83:387
Eo Y-J, Lee TH, Kang JK, Han YS, Bae B-S (2005) J Polym Sci B 43:827
Panitz J-C, Wokaun A (1997) J Sol–Gel Sci Technol 9:251
Gigant K, Posset U, Schottner G, Baia L, Kiefer W, Popp J (2003) J Sol–Gel Sci Technol 26:369
Gnyba M, Keranen M, Kozanecki M, Bogdanowicz R, Kosmowski BB, Wroczynski P (2002) Opto-Electron Rev 10:137
Klein LC (1985) Annu Rev Mater Sci 15:227
Acknowledgements
Dr. Jocelyne Galy (LMM-IMP-INSA) Lyon is gratefully acknowledged for the GPC measurements. MIUR is acknowledged for the financial support (PRIN 2005).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dirè, S., Tagliazucca, V., Brusatin, G. et al. Hybrid organic/inorganic materials for photonic applications via assembling of nanostructured molecular units. J Sol-Gel Sci Technol 48, 217–223 (2008). https://doi.org/10.1007/s10971-008-1752-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-008-1752-x