
Abstract
In situ silica reinforcement of natural rubber (NR) grafted with methyl methacrylate (MMA) (MMA-GNR) was achieved via the sol–gel reaction of tetraethoxysilane (TEOS) by the use of solid rubber and latex solutions. Silica contents within the MMA-GNR as high as 48 and 19 phr were obtained when using the solid rubber and latex solutions, respectively, under optimum conditions. The conversion efficiency of TEOS to silica was close to 95%. The in situ formed silica MMA-GNR/NR composite vulcanizates were prepared. MMA-GNR/NR composite vulcanizates reinforced with the in situ formed silica prepared by either method had similar mechanical properties to each other, but a shorter cure time and higher mechanical properties than those reinforced with the commercial silica at 9 phr. The TEM micrographs confirmed that the in situ formed silica particles were well dispersed within the MMA-GNR/NR composite matrix, whilst the commercial silica particles showed a significant level of agglomeration and a lower level of dispersion.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The modification of rubber by grafting with vinyl monomers has gained considerable interest and importance recently in modifying the properties of the rubber vulcanizates. Methyl methacrylate (MMA) and styrene have (currently) been found to be the most suitable monomers, when polymerized, to give a high level of grafting. MMA grafted natural rubber (NR) (MMA-GNR), with tert-butylhydroperoxide as the initiator, has been marketed since the mid 1950s under the trade name “Heveaplus” [1, 2]. The formation of MMA-GNR can simultaneously improve the physical properties and reduce the tackiness of the rubber film, and these composite particles of MMA-GNR generally have a ‘core–shell’ structure [2]. Although the particles are prepared in two consecutive or multistage emulsion polymerization steps, a core–shell structure does not necessarily occur. A number of other morphologies, designated as raspberry, sandwich and inverted core–shell, might be observed and several reaction parameters are known to affect the particle morphology. These include the compatibility of the monomer, surfactant concentration, monomer and polymer ratio, type of initiator used, temperature and degree of the grafting reaction [1–4]. However, the physical properties of the GNR vulcanizates can be greatly enhanced by reinforcement, yet novel studies on the reinforcement of GNR vulcanizates sadly seem to be rather scarce.
In the rubber industry, besides carbon black, silica is the other important reinforcing filler used to impart specific properties to rubber compounds. This is because the compounding of silica with NR (or other rubber compounds) offers a number of advantages on the vulcanizates, such as good tear strength, good abrasion resistance and reduction in the heat build-up [5]. Therefore, silica is used in products where a black color is not a requirement, e.g., in shoes soles. However, the use of conventional silica instead of carbon black for rubber reinforcement is limited due to the problems of silica imparting a higher compound viscosity due to the filler/filler interactions, with the resultant a greater difficulty in mixing and processing and a longer vulcanization time, with consequentially a lower network-chain density [6]. In order to overcome these problems, in situ polymerization (sol–gel process) of tetraethoxysilane (TEOS) in rubber has been performed. The sol–gel process enables the in situ formation of silica and its dispersion within the matrix, and so allows the silica to act as a good reinforcement agent in the rubber. The reaction of TEOS takes place in two steps, the hydrolysis and the condensation reactions, to produce silica (SiO2). This sol–gel process has been noted for the preparation of inorganic glasses at lower temperatures [7, 8].
Among several techniques, that of in situ silica formation in the polymer matrix is very simple and readily produces hybrid materials. The sol–gel process using a solid rubber method has been developed and applied to polymers such as styrene-butadiene (SBR) [9], epoxidized natural rubber [10, 11] and NR [12–16]. As already noted, the sol–gel reaction of TEOS to form in situ silica produces fine and well-dispersed silica particles in the rubber matrix. Alternatively, the sol–gel process can be performed using a latex solution, where the TEOS is directly added into the rubber latex and thus is present during the grafting process, rather than after it, as is the case for the solid rubber method. Both methods are followed by heat curing to convert the TEOS to silica and to form the rubber composite. The reaction of TEOS to silica has been reported to go to completion in latexes, such as acrylonitrile-butadiene rubber (NBR) [17], SBR [17], PMMA [18–20], NR [21, 22] and GNR [23]. However, with the sol–gel process using a rubber latex solution, the amount of in situ silica produced and the reinforcing properties of the rubber were found to be dependent on the amount of TEOS present, the reaction temperature and time [17]. The amount of TEOS added and the [H2O]/[TEOS] molar ratio were both found to be related to the amount of in situ silica produced, the silica particle size and the reinforcing behavior of the silica in both SBR and NBR [17]. The mechanical properties of the rubber vulcanizates were also affected by the amount of TEOS added into the latex solution and the small amount of ammonia added as a catalyst for the sol–gel reaction of NR [21]. For the sol–gel reaction of MMA-GNR, the in situ formed silica particles were found to be dispersed in the GNR matrix and to slightly increase both the modulus and the tear strength of the MMA-GNR composites. However, the in situ silica introduced by the sol–gel technique using the solid rubber and latex solution methods both yielded a better reinforcing efficiency than the commercial silica.
When generated in situ, silica is the most interesting nanofiller produced in the rubber matrix and the effect of silica dispersion on reinforcement has been widely investigated [9–12, 14–17]. Another interesting point is that the sol–gel process using the latex solution method does not require the addition of a catalyst since the presence of the NH3 preserver in the latex acts as a catalyst [17, 21], whereas the solid method requires the addition of an amine catalyst for the sol–gel reaction. The aim of this work was to produce in situ formed silica reinforced MMA-GNR using both the latex and solid rubber methods. The mechanical properties of the resultant in situ formed silica reinforced MMA-GNR/NR composite vulcanizates were then investigated and compared with each other and with that reinforced with commercial silica.
2 Experimental
2.1 Materials
The high ammonia NR latex with a dry rubber content of 60.0% was obtained from the Thai Rubber Latex Corp. (Thailand) Public Co. Ltd. MMA (Merck, AR) was washed with 10% (w/v) aqueous sodium hydroxide to remove the inhibitor, dried over anhydrous magnesium sulfate and stored at 0 °C. Sodium dodecyl sulphonate (SDS) (Fisher Scientific), cumene hydroperoxide ammonia solution (CHPO) (Merck, AR), tetraethylene pentamine (TEPA) (Merck, AR), TEOS (Fluka, AR) and n-hexylamine (Aldrich, AR) were used as received. Zinc oxide (Pan Innovation Ltd.), stearic acid (Imperial Industrial Chemicals Co. Ltd.), polyethylene glycol (Pan Innovation Ltd.), mercaptobenzothiazole disulfide (MBTS) (Pan Innovation Ltd.) were used for vulcanization. The commercial silica was supplied by PPG-Siam Silica Co., Ltd. The solid NR (STR5L) was provided by Thai Hua Chumporn Natural Rubber Co., Ltd. (Thailand).
2.2 Sol–gel reaction of MMA-GNR using the solid rubber method
NR latex (50 g) was added into a round-bottom reactor along with 100 mL deionized water. Potassium hydroxide solution and SDS at 1 phr final concentration, based on the dry rubber content were added, while stirring. The mixture was deoxygenated by purging with nitrogen gas for approximately 15 min at room temperature and then oleic acid (10 phr) was added with stirring, followed by the addition of the MMA monomer over 30 min with stirring to allow the the latex particles to swell. The mixture was then heated up to 50 °C whereupon the bipolar redox initiating system (CHPO: TEPA molar ratio of 1:1) was added to 1 phr final concentration. The reaction was allowed to proceed for 8 h under continuous stirring to complete the polymerization. The final MMA-GNR was coagulated using ethanol. The conversion of graft copolymerization was determined by the percentage increase in the rubber weight.
The amount of MMA-GNR, free NR and free PMMA in the product was determined by soxhlet extraction. The free NR was extracted by light petroleum ether (60–80 °C) for 24 h. The residue was dried to a constant weight in an oven at 40 °C under a vacuum for 24 h. To remove the free PMMA, the residue was extracted in acetone. The weight difference between the initial sample and extracted samples was used to evaluate the free NR, free PMMA and GNR levels and the grafting efficiency, from the following equations.
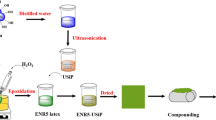
The solid MMA-GNR was prepared into thin sheets of ca. 1 mm thickness on a two-roll mill and the sheets were then immersed in TEOS solution (20–70 phr) at 30–40 °C for 1–24 h in a container. After that, the swollen sheets were immersed in an aqueous solution of n-hexylamine (8 or 64 mM) as the catalyst and finally dried under vacuum at 40 °C. The preparation procedure is schematically summarized in Fig. 1.

Schematic summary of the preparation of in situ formed silica reinforced MMA-GNR using the solid rubber (InSi9-S) and liquid latex solution (InSi9-L) methods
2.3 Sol–gel reaction of MMA-GNR using the latex solution method
The MMA-GNR was reinforced with silica by the sol–gel process using TEOS in the latex solution. Two methods of adding the TEOS into the sol–gel reaction were studied. In the first method the TEOS is added during the graft copolymerization reaction, whereas in the second method the TEOS is added after the graft copolymerization. The preparation procedure is summarized in Fig. 1. For the first method, the TEOS was added with the MMA monomer and the graft copolymerization and sol–gel reaction proceeded simultaneously at 50–60 °C for 8 h in the presence of the CHPO: TEPA (1:1 molar ratio) initiator. For the second method, the MMA-GNR latex was prepared by graft copolymerization at 50 °C for 8 h, also using 1 phr of CHPO: TEPA (1:1 molar ratio) as the initiator, but the TEOS was then added into the grafted rubber latex and the sol–gel reaction was carried out at 50–60 °C for 1–5 days.
2.4 Preparation of the MMA-GNR/NR vulcanizates
The in situ formed silica grafted rubber sheet with a silica content of either 18 phr or 40 phr was used as the starting material and diluted by the addition of virgin NR. The compound formulae of the MMA-GNR/NR blends were as follows: MMA-GNR/NR = 50/50; in situ formed silica filled MMA-GNR/NR = 59/50 (for 9 phr silica); in situ formed silica filled MMA-GNR/NR = 70/50 (for 20 phr silica), all with 5 phr zinc oxide, 2 phr stearic acid, 1.8 phr MBTS, 3 phr sulfur and 0.4–0.8 phr PEG. The NR was first masticated using a two-roll mill at room temperature before mixing with the in situ formed silica reinforced MMA-GNR or silica free MMA-GNR, as appropriate, and then the additives were then added. The MMA-GNR/NR composite blends (with or without silica filling) were vulcanized in a compression mold at 150 °C for a specific cure time, determined using a rheometer.
2.5 Characterization
The functional groups of the NR and MMA-GNR were characterized using Fourier Transform Infrared Spectroscopy (FT-IR; Bruker 3,000 × spectrometer). Each sample was dissolved in chloroform and cast on a KBr cell. The FT-IR spectra were determined in range of 400–4,000 cm−1 with 32 scans at a resolution of 4 cm−1.
The chemical structure of the MMA-GNR was confirmed using 1H-NMR spectrometry. Samples were dissolved in CDCl3 and then the 1H-NMR spectrum was taken using a Varian NMR spectrometer operating at a proton resonance frequency of 400 MHz.
The amount of silica content was determined by using thermogravimetric analysis (TGA) on a TG/DTA Perkin–Elmer machine to determine the amount of in situ formed silica generated in the MMA-GNR. Each MMA-GNR sample (ca.10 mg) was placed in a platinum pan and heated under air at room temperature up to 1,000 °C using a heating rate of 10 °C/min under a nitrogen gas flow rate of 50 mL/min. The silica content and the conversion of TEOS to silica were calculated as follows:
where W1 is the weight of remaining ash and W2 is the composite weight.
where W3 is the amount of in situ generated silica in the sample, obtained from eq. (5) and W4 is the theoretical amount of silica being generated assuming a quantitative conversion of TEOS to silica by the overall reaction as follows:
The dispersion and particle size of the silica filler in each vulcanizate was studied using transmission electron microscopy (TEM). Ultra thin films of the vulcanized rubber sample were prepared using a microtome (Boeckeler Instrument, Inc.) in liquid nitrogen, placed on a copper grid and then coated by carbon. The samples were then observed with a transmission electron microscope (JEOL JEM-2010 instrument, Hitachi Co. Ltd.) operating at an accelerating voltage of 200 kV.
2.6 Mechanical properties of the vulcanizates
The tensile properties of the samples were measured according to ASTM D412, and performed on a Universal testing machine (LLOYD LR 10 K) with a crosshead speed of 500 mm/min, and a load cell of 5 kN. Values reported for each sample were based on an average of five measurements. The tear strength was measured according to ASTM D624-02, and performed on a Hounsfield H10KS testing machine at a crosshead speed of 500 mm/min. The hardness of the sample was measured according to ASTM D2240 using a Shore-type-A Lever Loader (REX GAUGE 2000 & OS-2 Stand).
The abrasion resistance of the sample was measured using a Happen Abrasion machine (ZWICK) according to DIN 53516. The values are reported on the “volume loss” basis, calculated from the weight loss and density of the compound.
3 Results and discussion
3.1 Graft copolymerization of MMA onto NR latex
The graft copolymer of MMA onto NR was prepared by emulsion polymerization with the CHPO/TEPA redox system as the initiator at 50 °C for 8 h. The monomer conversion level obtained was 66.7% with a grafting efficiency of 39.2%. The yield of GNR obtained was 51.7%, whilst the free PMMA and NR levels were 19.6 and 28.7%, respectively.
The MMA-GNR was characterized by FT-IR and NMR spectroscopy. Characteristic FT-IR transmission peaks for NR at 2,958, 1,442, 1,371 and 837 cm−1, which correspond to the stretching vibration of aliphatic = C–H, C–H bending of CH2, C–H bending of CH3 and C = C bending vibration, respectively, were observed (Fig. 2). The evidence for MMA being present in the MMA-GNR graft copolymer was observed at 1,728 cm−1 for C = O stretching and 1,142 cm−1 for the –C–O—moiety of the ester functional groups of MMA. The 1H-NMR spectrum of the MMA-GNR revealed peaks at 1.67, 2.01 and 5.12 ppm that correspond to the NR, whilst the signal at 3.69 ppm corresponds to the methoxy group of MMA (Fig. 3). Together, these results from the FT-IR and 1H-NMR analyses indicate that MMA was grafted onto the NR.

FT-IR spectra of the NR and MMA-GNR

1H-NMR spectrum of the MMA-GNR
3.2 Sol–gel reaction of grafted rubber using the solid rubber method
3.2.1 Swelling degree of grafted rubber
The MMA-GNR sheets were immersed in TEOS at various times (range from 1 to 32 h) at either 40 °C or at room temperature (30 °C). The degree of swelling was calculated from the weight of the rubber sample before and after immersion, according to ASTM D471. Figure 4 shows the effect of temperature and immersing time on the degree of swelling of MMA-GNR sheets in TEOS. For immersion at 40 °C, the swelling degree of the MMA-GNR sheets sharply increased to 120% in the first hour but then only slightly increased over the next 23 h to a final swelling degree of 139%. This is likely to be due to the limiting diffusion (mass transfer limitation). For immersion at 30 °C, the swelling degree of MMA-GNR increased more slowly, compared to that at 40 °C, with the swelling plateau being approached at 24–32 h but attaining a higher final swelling level of ~150%. Thus, in order to keep a short time for the sol–gel process (for example for commercial economic viability), the sequential immersion of grafted rubber in TEOS at 40 °C for 1 h and 30 °C for 24 h was carried out where the highest swelling degree of 150% could still be achieved. The swollen MMA-GNR sheets were then further immersed in catalyst solution as detailed below.

The degree of swelling of the MMA-GNR in TEOS at 30 and 40 °C
3.2.2 Effect of the n-hexylamine (Catalyst) concentration
The swollen MMA-GNR sheet was immersed in an aqueous solution of either 8 or 64 mM n-hexylamine at 50 °C as the catalyst. Increasing the catalyst concentration from 8 to 64 mM slightly increased both the initial rate and the total amount of the in situ generated silica in the MMA-GNR over the 5-day reaction period (Fig. 5). For the solid rubber method, the amino group of the n-hexylamine catalytically promoted the sol–gel reaction of TEOS to generate in situ silica particles, attaining a yield as high as 48.7 phr silica and a conversion efficiency of TEOS to silica of about 94.8% with 64 mM n-hexylamine. The primary alkylamines with long hydrocarbon segments are assumed to form reverse micelles like a surfactant in the TEOS-swollen NR matrix [24, 25]. That the in situ formed silica content increased when the catalyst concentration was increased has been reported previously for the in situ silica generation in NR [15].

Effect of the n-hexylamine catalyst concentration on the sol–gel reaction using the solid rubber method at 50 °C a silica content (phr) and b the TEOS conversion efficiency (%) at 64 mM n-hexylamine
3.2.3 Effect of reaction temperature and time
When the swollen MMA-GNR sheet was immersed in aqueous solution of 64 mM n-hexylamine at 40 °C, 50 °C or 60 °C, the rate of formation and the total yield of the in situ formed silica was clearly temperature dependent, increasing as the reaction temperature increased (Fig. 6). At 40 and 50 °C, the in situ formed silica yield approached a maximum value after 3 days and remained constant beyond this. In some contrast, at 60 °C the initial rate and maximum yield of in situ formed silica produced was significantly higher, with the maximal yield being obtained from 2 days onwards. The final in situ formed silica content in the MMA-GNR matrix thus increased with increasing reaction temperature (at least within the range of 40–60 °C). According to the Arrhenius equation, the sol–gel reaction proceeded at a higher rate at the higher temperatures, with the optimum sol–gel reaction using the solid rubber method being obtained at 60 °C for 5 days, yielding an in situ silica content of 48 phr and a conversion efficiency of 94%.

Effect of the reaction temperature and time on the sol–gel reaction using the solid rubber method a silica content (phr) and b the TEOS conversion efficiency (%) at 64 mM n-hexylamine
3.3 Sol–gel reaction of grafted rubber using the latex solution method
3.3.1 Effect of TEOS addition during or after graft copolymerization
A comparison of the sol–gel reaction when the TEOS was either added during or else after the grafting reaction was evaluated. The in situ formed silica content and the conversion efficiency (%) obtained in the MMA-GNR when the TEOS (20 phr) was added during and after the graft copolymerization is summarized in Fig. 7. At 50 °C the in situ formed silica yield increased with an increasing reaction time and approached a constant (maximal) value after 3 days when the TEOS was added during the grafting reaction. However, when the TEOS was added after the grafting reaction an initial slower in situ silica formation rate was observed over the first 3 days, followed by (after almost no increase from the third to the fourth day) a rapid increase in the production rate to tend towards a plateau at 5–6 days and reach a maximal yield after 6–7 days of reaction time. The maximal yield of in situ formed silica (~5.5 phr) and the conversion efficiency of TEOS into silica (>90%) were broadly similar between the two methods.

Effect of addition method of TEOS (20 phr) on the sol–gel reaction using the latex solution method a the silica content (phr) and b the TEOS conversion efficiency (%) at different reaction times
When the reaction was performed at 60 °C and the TEOS was added during the grafting, a slightly faster initial kinetics of in situ formed silica production was noted compared to that at 50 °C, with the maximal yield plateau being reached sooner, but the overall maximal in situ formed silica yield and TEOS conversion efficiency were not significantly different.
Interestingly, the total yield of MMA-GNR formed when the TEOS was added after the graft reaction was 1.32-fold higher than that obtained when the TEOS was added during the graft reaction (Fig. 8). Correspondingly, lower levels of free NR (~1.2-fold) and free PMMA (~1.16-fold) were observed when TEOS was added after the grafting reaction. Perhaps when the TEOS was added during the grafting reaction, the silica particles in MMA-GNR latex could retard or inhibit the grafting reaction between the MMA monomer and the NR chain. Given that, additionally, the latex solution tends to coagulate when TEOS was added at concentrations above 20 phr, whereas when the TEOS was added after the grafting reaction the latex solution was stable allowing TEOS loadings of up to 70 phr, then the addition of the TEOS after the graft copolymerization is more optimal than during it.

Effect of addition method of TEOS (20 phr) on the proportions of the GNR formed, along with residual levels of free (ungrafted) NR and PMMA, in the in situ formed silica reinforced MMA-GNR
For the sol–gel reaction with the addition of TEOS after grafting, the effect of the TEOS concentration on the yield of in situ formed silica and the TEOS conversion efficiency (%) are presented in Fig. 9. Note that the MMA-GNR latex coagulated when TEOS concentrations above 70 phr were added, and so forms the upper limit. The in situ formed silica content increased with increasing TEOS concentrations in the 10–70 phr range, where the TEOS can undergo hydrolysis and condensation to form silica. A TEOS conversion efficiency of up to 90% was also observed. The optimum condition of the sol–gel reaction using the latex method was at a TEOS concentration of 70 phr and a temperature of 50 °C, yielding a maximum in situ formed silica content of 17.9 phr. The TEOS conversion to silica also increased with time and approached 89–90% for all TEOS concentrations in the tested 10–70 phr range, with higher TEOS concentrations giving a higher initial rate of conversion over the first 1–2 days of the reaction. This is likely to reflect the higher diffusion rates at higher TEOS concentrations. Therefore, from 2 to 7 days, the amount of TEOS did not significantly affect the final TEOS conversion level obtained. Under these conditions most of the TEOS molecules had produced silica particles in the MMA-GNR matrix according to the hydrolysis and condensation reaction [21].

Effect of the TEOS concentration on the sol–gel reaction using the latex solution method (TEOS addition after grafting) at 50 °C a in situ formed silica content (phr) and b the TEOS conversion efficiency (%) at different reaction times
3.4 Morphology of the MMA-GNR/NR composite vulcanizates
The morphology of the silica reinforced MMA-GNR/NR composite vulcanizates were characterized by TEM analysis of thin sections. The surface morphology of the in situ formed silica MMA-GNR/NR composites prepared by the sol–gel process using either the latex solution or the solid rubber methods with a silica level of 9 phr or 20 phr are shown in Fig. 10, along with that produced with commercial silica particles at 9 phr. The MMA-GNR/NR composite vulcanizate produced with the commercial silica particles at 9 phr showed that the silica was aggregated to form agglomerated structures, no doubt due to the highly polar and hydrophilic surfaces of the precipitated silica and the presence of numerous silanol groups allowing the silica particles to agglomerate easily [22]. On the other hand, the in situ formed silica particles produced as a result of either the latex solution or the solid rubber methods were dispersed more homogeneously in the NR matrix, and so the agglomeration structures of the in situ formed silica particles are much smaller than that of the commercial silica particles. The morphology of the in situ formed silica in the MMA-GNR/NR composite prepared by using either the solid rubber or the latex solution methods were not significantly different. When the in situ formed silica loading was increased from 9 to 20 phr, the level of agglomeration of the in situ formed silica particles increased. The schematic representation of the in situ silica generated in the GNR matrix is shown in Fig. 11. Here the silica particles dispersed in the GNR interact with each other more than with the rubber molecules (Fig. 11a). On the other hand, the carbonyl groups of the MMA molecules were bonded to the silanol groups of the silica particles to produce hydrogen bonds (Fig. 11b). Hence, the dispersion of silica in the MMA-GNR matrix was more homogeneous than that of the silica dispersed in NR.

TEM photographs of the MMA-GNR/NR composite vulcanizates for the a commercial silica at 9 phr and (b–d) in situ formed silica formed via the (b) latex solution method (9 phr) or (c, d) via the solid rubber method at (c) 9 phr or (d) 20 phr

Schematic representation of silica particles dispersed in the rubbery matrix of (a) NR and (b) MMA-GNR
3.5 Mechanical properties of the different MMA-GNR/NR composite vulcanizates
The MMA-GNR/NR composite vulcanizates, with or without reinforcing with either commercial or in situ formed silica particles were prepared and then their mechanical properties, in terms of their tensile strength, elongation at break, 300% modulus (M300), tear resistance, hardness and abrasion resistance, were evaluated. The results are summarized in Table 1. Comparing the tensile properties of NR and the MMA-GNR/NR composite vulcanizates (no silica), it was found that the 300% modulus and the tensile strength of MMA-GNR/NR composite vulcanizates were significantly (4.9- and 1.2-fold, respectively) higher than that of the NR vulcanizate, which is likely to be due to hard segmented PMMA molecules in the NR chain [26]. The elongation at break of the MMA-GNR/NR composite vulcanizates were found to be 1.2-fold lower than that of the NR vulcanizate, again likely to be due to the increasing chain stiffness of the PMMA in the NR.
Comparing the mechanical properties of the MMA-GNR/NR composite blends reinforced with silica particles at 9 phr, it was clear that the inclusion of the silica particles further increased the tensile strength and the 300% modulus, and decreased the elongation at break. However, whilst the MMA-GNR/NR composite vulcanizates reinforced with the in situ silica (9 phr) formed by either the liquid latex or solid rubber methods showed a broadly similar tensile strength, 300% modulus and elongation at break values, those with commercial silica at 9 phr showed a lower tensile strength and 300% modulus, and a higher elongation at break. This is likely to be due to the fact that the in situ formed silica generated by either method is homogenously dispersed throughout the MMA-GNR matrix, which contributed effectively to the reinforcement, in contrast to the agglomerated commercial silica particles, as seen in the TEM micrographs (Fig. 10). Likewise, the 300% modulus and tensile strength increased, and the elongation at break decreased, when the in situ formed silica content in the reinforced MMA-GNR/NR composite vulcanizates were increased from 9 to 20 phr, because the stress applied from the external force to the composite was transferred to the load-bearing filler (which were the load-bearing element) through the filler-matrix interface [8].
The hardness of the MMA-GNR/NR composite vulcanizate (no silica) was 1.6-fold higher than that of the NR vulcanizate, and this increased a further 1.08- to 1.09-fold with the inclusion of silica at 9 phr, but was essentially the same when reinforced by the commercial or either of the in situ formed silica preparations at the same level (9 phr). Increasing the in situ formed silica content from 9 to 20 phr, however, resulted in a ~1.07-fold further increase in the hardness of the resultant silica reinforced MMA-GNR/NR composite vulcanizate.
The tear strength of the MMA-GNR/NR composite vulcanizate (no silica) was some 1.7-fold higher than that of NR vulcanizate, again likely to be due to the presence of the hard segments of PMMA in the MMA-GNR. The tear strength was further increased (1.07- to 1.13-fold) upon the inclusion of silica at 9 phr, but the tear strength of the two vulcanizates with the different in situ formed silica preparations, although similar to each other, were slightly higher than that for the vulcanizate reinforced with commercial silica at the same level. The highest tear strength was, however, seen with the inclusion of the higher (20 phr) level of in situ formed silica. This can be explained as that when the size of the agglomerated silica particles was reduced, a good bonding between the silica particles and the network so formed reduced the load transfer between them [23].
Finally, the abrasion resistance, as measured by the volume loss of the compound after applying an external force, was slightly (1.07-fold) increased in the MMA-GNR/NR composite vulcanizate (no silica) compared to the NR vulcanizate, and was further increased following the inclusion of silica at 9 phr from the commercial (1.12-fold higher) or with the in situ formed silica by both methods (1.23- to 1.22-fold). This is also likely to be due to the fact that the commercial silica was not well dispersed in the matrix and had a higher agglomeration, as supported by the TEM micrographs (Fig. 10). Increasing the in situ formed silica content in the MMA-GNR/NR composite vulcanizates from 9 to 20 phr also increased the abrasion loss ~1.12- to 1.13-fold.
Overall, the greatest changes in the mechanical properties of the NR vulcanizates were seen with the inclusion of the in situ formed silica into the MMA-GNR/NR composite at 20 phr, followed by those MMA-GNR vulcanizates reinforced with 9 phr of in situ formed silica, which were more effective than the inclusion of the commercial silica at the same level.
3.6 Thermal stability of the NR/GNR vulcanizates
The thermal stability and the decomposition behavior of vulcanizates were investigated using TGA in the presence of a nitrogen atmosphere. The thermal decomposition of NR was a one-stage reaction (Fig. 12) with a single decomposition temperature range of 308–373 °C (Table 2). The differential thermogravimetric (DTG) curves of silica reinforced GNR/NR vulcanizates with or without silica incorporation showed a two-stage decomposition process, with the first decomposition stage having peak temperatures of about 306–316 °C (Tid) and a range of 370–374 °C (Tmax,1), and corresponded to the decomposition of the soft segment (polyisoprene) of the MMA-GNR. The second thermal decomposition stage occurred at a temperature of about 403–406 °C and was attributed to the decomposition of the hard PMMA segment of the MMA-GNR. Thus, the graft chain of NR promoted the stability of the vulcanizates. However, the thermal stability and the decomposition behavior did not change with increasing the level of the in situ formed silica from 9 to 20 phr. Rather, the in situ formed silica reinforced vulcanizates (9 and 20 phr) and the commercial silica reinforced vulcanizate (9 phr) all had a similar thermal stability.

TGA thermograms of the in situ silica reinforced MMA-GNR/NR composite vulcanizates including those reinforced with in situ formed silica produced by the latex solution method at 9 phr silica (Si9-L) or by the solid rubber method at either 9 (Si9-S) or 20 (Si20-S) phr silica
4 Conclusion
The graft copolymerization of MMA onto NR was carried out using CHPO/TEPA as the initiator. The structure of the MMA-GNR was confirmed by FT-IR and NMR analysis. The in situ formed silica reinforced MMA-GNR composite was prepared by a sol–gel process via TEOS using a solid rubber method with n-hexylamine as the catalyst. The yield of in situ formed silica increased as the concentration of n-hexylamine, the TEOS content or the reaction temperature and time were increased. For the latex solution method, the in situ formed silica content increased as the TEOS concentration, reaction temperature and time were increased. A lower percentage of GNR was found in the in situ formed silica reinforced MMA-GNR when the TEOS was added during the graft copolymerization than when added after the graft copolymerization. The optimum condition of the sol–gel process using the solid rubber method was n-hexylamine concentration of 64 mM at a temperature of 60 °C, which achieved the relatively high silica content of 48 phr and a TEOS conversion level of almost 95%. For the sol–gel process using the latex solution method, the optimum condition was with 70 phr TEOS and 50 °C, which resulted in silica content of 18 phr. The mechanical properties of the in situ formed silica reinforced MMA-GNR/NR composite vulcanizates at 9 phr silica were higher than that for the commercial silica filled vulcanizates at the same 9 phr silica, but the mechanical properties were further increased when the in situ formed silica content was increased from 9 to 20 phr in the MMA-GNR/NR composite vulcanizates. The TEM micrographs confirmed that the in situ formed silica particles were well dispersed in the GNR matrix whereas a higher level of silica particle agglomeration was seen for the commercial silica particles in the GNR matrix.
References
Muroi S, Hashimoto H, Hosoi K (1984) Morphology of core-shell latex particles. J Appl Polym Sci 22:1365–1372
Merkel MP, Domonie V, El-Aasser MS, Vanderhoff JW (1987) Process parameters and their effect on grafting reactions in core/shell latexes. J Appl Polym Sci 25:1755–1767
Dimonie V, El-Aasser MS, Vanderhoff JW (1984) Core-shell emulsion copolymerization of styrene and acrylonitrile on polystyrene seed particles. J Polym Sci Part A-1 Polym Chem 22:2197–2215
Kochthongrasamee T, Prasassarakich P, Kiatkamjornwong S (2006) Effect of redox initiator on graft copolymerization of methyl methacrylate onto natural rubber. J Appl Polym Sci 101:2587–2601
Schwaiger B, Blume A (2000) Silica/silane a winning formula in reinforcement. Rubber World 222:32–38
Brinker CJ, Scherer GW (1990) Sol-gel science: The Physics and Chemistry of Sol-gel Processing. Academic Press, New York
Mark JE (1985) Science of ceramic chemical processing. Wiley, New York, Chapter 47
Kohjiya S, Ikeda Y (2000) Reinforcement of general-purpose grade rubbers by silica generated in situ. Rubber Chem Technol 73:534–550
Ikeda Y, Tanaka A, Kohjiya S (1997) Effect of catalyst on in situ silica reinforcement of styrene–butadiene rubber vulcanizate by the sol–gel reaction of tetraethoxysilane. J Mater Chem 7:445–458
Hashim A, Kawabata N, Kohjiya S (1995) Silica reinforcement of epoxidized natural rubber by sol-gel method. J Sol-Gel Sci Technol 5:211–218
Bandyopadhyay A, Sarkar MD, Bhowmick AK (2005) Epoxidised natural rubber/silica hybrid nanocomposites by sol-gel technique: effect of reactants on the structure and the properties. J Mater Sci 40:53–62
Ikeda Y, Kohjiya S (1997) In situ formed silica particles in rubber vulcanizate by the sol-gel method. Polymer 38:4417–4423
Ikeda Y, Katoh A, Shimanuki J, Kohjiya S (2004) Nano-structural observation of in situ silica in natural rubber matrix by three dimensional transmission electron microscopy. Macromol Rapid Commun 25:1186–1190
Poompradub S, Kohjiya S, Ikeda Y (2005) Natural rubber/in situ silica nanocomposite of a high silica content. Chem Lett 34:672
Ikeda Y, Poompradub S, Kohjiya S, Morita Y (2008) Preparation of high performance nanocomposite elastomer: effect of reaction conditions on in situ silica generation of high content in natural rubber. J Sol-Gel Sci Technol 34:299–306
Chaichua B, Prasassarakich P, Poompradub S (2009) In situ silica reinforcement of natural rubber by sol-gel reaction via rubber solution. J Sol Gel Sci. Tech. 52:219–306
Yoshikai K, Ohsaki T, Furukawa M (2002) Silica reinforcement of synthetic diene rubbers by sol–gel process in the latex. J Appl Polym Sci 85:2053–2063
Landry CJ, Contrain KB, Brady BK (1992) In situ polymerization of tetraethoxysilane in poly (methyl methacrylate): morphology and dynamic mechanical properties. Polymer 33:1486–1506
Silveira FK, Yoshida VP, Nunes PS (1995) Phase separation in PMMA/silica sol-gel systems. Polymer 36:1425–1434
Zulficar AM, Mohammad WA, Kadhum AA, Hilal N (2007) Synthesis and characterization of poly(methyl methacrylate)/SiO2 hybrid membrane. J Mater Sci 45:422–426
Tangpasuthadol V, Intasiri A, Nuntivanich D, Niyompanich N, Kiatkamjornwong S (2008) Silica-reinforced natural rubber prepared by the sol–gel process of ethoxysilanes in rubber latex. J Appl Polym Sci 109:424–433
Xu H, Liu J, Fang L, Wu C (2007) In situ grafting onto silica surface with epoxidized natural rubber via solid state method. J Macromol Sci 46:693–703
Satraphan P, Intasiri A, Tangpasuthadol V, Kiatkamjornwong S (2009) Effects of methyl methacrylate grafting and in situ silica particle formation on the morphology and mechanical properties of natural rubber composite films. Polym Adv Technol 20:473–486
Osseo AK, Arriagada FJ (1999) Growth kinetics of nanosize silica in a nonionic water-in-oil microemulsion: a reverse micellar pseudophase reaction model. J Colloid Interface Sci 218:68–76
Debuigne F, Jeunieau L, Wiame M, Nagy JB (2000) Synthesis of organic nanoparticles in different W/O microemulsions. Langmuir 16:7605–7611
Thiraphattaraphun L, Kiatkamjornwong S, Prasassarakich P, Damronglerd S (2001) Natural rubber-g-methyl methacrylate/poly(methyl methacrylate) blends. J Appl Polym Sci 81:428–439
Acknowledgments
The funding support from Toyo Tire & Rubber Co. Ltd. (Japan), the Thai Government Stimulus Package 2 (TKK2555) under the Project for Establishment of Comprehensive Center for Innovative Food, Health Products and Agriculture, the National Research University Project of CHE and the Ratchadaphiseksomphot Endowment Fund (AM1024I) are gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Watcharakul, N., Poompradub, S. & Prasassarakich, P. In situ silica reinforcement of methyl methacrylate grafted natural rubber by sol–gel process. J Sol-Gel Sci Technol 58, 407–418 (2011). https://doi.org/10.1007/s10971-011-2407-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10971-011-2407-x