
Abstract
Plutonium is one of the key radionuclides in nuclear decommission. In this study, a rapid method was developed to analyze Pu for concrete samples using total digestion, CaF2/LaF3 coprecipitation, extraction chromatography using TEVA and DGA resins, and ICP-MS measurement. The whole analytical process can be achieved within 2 days, with sufficiently high decontamination factors of interfering elements and high Pu chemical recovery (57–93%). The high throughput (22 samples/2 days) and low LODs (0.008 mBq g−1 for 239Pu and 0.02 mBq g−1 for 240Pu) allow this method to effectively detect low level Pu contamination in concrete samples.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
In the decommission of a nuclear facility or a nuclear accident site, various types of building materials need to be disposed, e.g. bio-shielding concrete and steel waste. Before the establishment of an appropriate disposal strategy, a field investigation should be conducted to assess the inventory and the corresponding distribution of the target radionuclides, including fission products and actinides. In contrast to the short-lived fission products which can be determined directly by radiometric measurement, the determination of actinides is more challenging due to their long half-lives and relatively low concentrations, especially for transuranium nuclides. Plutonium is one of the key nuclides in decommission not only due to its radioactivity and toxicity, but also due to the long half-lives which enable its presence after thousands of years since its burial. In the bio-shielding concrete material which typically contains μg g−1 level uranium, 239Pu can be formed via the following nuclear reaction [1]:
It is thus essential to assess the concentration of Pu before the disposal of concrete waste. In the literature, dozens of methods have been reported to analyze Pu isotopes for soil and sediment [2,3,4,5,6,7,8]. However, these methods are mainly intended for environmental Pu originated from global fallout. In the case of determining Pu for decommission concrete samples; the requirements on chemical separation are somewhat different. Firstly, the Pu activity in decommission concrete sample is much higher than environmental level and smaller sample size can be used for analysis. Secondly, Pu in the concrete might be in the form of silicate or hot particles and a complete digestion of the sample is required. Thirdly, hundreds of samples need to be analyzed to obtain a full picture of its distribution; a method with high throughput and short turnaround time is preferred. These reported methods for environmental Pu analysis might not be employed directly to analyze Pu for decommission concrete. In the literature, however, only several methods were specially developed to analyze Pu in concrete [1, 9, 10]. For example, Weinreich et al. [1]. reported a method for concrete by combining Pu dissolution using HNO3–HF–H3BO3 method, Pu separation using Biorad 1 × 2 resin and Eichrom UTEVA resin, Pu measurement using alpha spectrometry. Qiao and Hou [10] reported a method to determine Pu fractionations in concrete samples by sequential extraction procedure using various reagents, which were subsequently separated by ionic exchange chromatography and measured by alpha spectrometry. Maxwell et al. [9] reported a simultaneous method to determine actinides in concrete and brick samples by employing NaOH fusion, LaF3 coprecipitation, extraction chromatographic separation using Eichrom TEVA, TRU and DGA resins and alpha spectrometric measurement. It should be noted that all above mentioned methods used alpha spectrometry for Pu measurement, which usually takes days for measurement. Although their chemical separation was rapid, the long counting time decreased the data acquisition efficiency.
In this study, attempt was made to develop a rapid method to analyze Pu in decommission concrete samples by inductively coupled plasma mass spectrometry (ICP-MS) which is known for its high sensitivity and short measurement time. Due to the different characteristics of alpha spectrometry and ICP-MS, the sample preparation processes for both techniques are different. For example, the chemical separation for ICP-MS measurement should remove elements such as U, Pb, Tl, Pt, Hg and Bi, to eliminate the spectral interferences on Pu determination [11]. While the interfering elements for the determination of Pu using alpha spectrometry are Am, Po, Th, Ra, Pa and U [12]. In addition, elution reagents of Pu in ionic or extraction chromatography for alpha spectrometry which contain metallic reduction reagents (e.g. Fe2+ and Ti3+) might not be compatible with ICP-MS measurement [11]. In present study, on a basis of our previous reported method for the determination of Pu in soil and sediment by ICP-MS [11], modifications were made to meet the requirements in determining Pu isotopes for decommission concrete samples. After the evaluation by analyzing two quality control materials IAEA-384 and IAEA-385, the developed method was applied to analyze actual decommission concrete samples.
Experimental
Instrumentation
A milling machine (Pulverisette 5, Fritsch, Germany) was employed to crush the concrete samples. A centrifuge (Avanti J-E, Beckman Coulter, IN, USA) was utilized to achieve coprecipitation. The extraction chromatographic separation was conducted on a 24-hole polycarbonate vacuum box (Eichrom, IL, USA) which was coupled with a vacuum pump (N820.3 FT.18, KNF, Freiburg, Germany). Pu isotopes were determined by a sector field ICP-MS (Element XR, Thermo Scientific, Bremen, Germany) which used a scott type spray chamber as the sample introduction system. For the Pu analysis of low level concrete samples (e.g. samples analyzed in this study), the ICP-MS instrument can be placed in an open space. However, for the analysis of concrete samples with high level Pu contamination, it is suggested that the sample introduction apparatus and waste solution container of the ICP-MS instrument should be contained in fume hood, from the aspect of radiation protection.
Reagents and materials
Analytical grade reagents including HNO3, HCl, HF, HClO4, H3BO3, Ca(NO3)2·4H2O, La(NO3)3·6H2O, TiCl3, NaNO2, NH2OH·HCl and ascorbic acid were purchased from Aladdin Biochemical Technology (Shanghai, China). Iron (II) sulfamate (Fe(NH2SO3)2) was obtained from Strem Chemicals (MA, USA). Deionized water (18 MΩ cm) was used in the preparation of solutions. UTEVA, TEVA and DGA extraction resins were purchased from Triskem International (Bruz, France), and all resins were in the form of 2 mL cartridge with 50–100 μm particle size. 233U (Institute for Reference Materials and Measurements, Belgium) and 242Pu (Eckert and Ziegler Isotope Products, USA) were used in this study as internal standard and chemical tracer respectively. Standard reference materials (SRMs) including IAEA-384 and IAEA-385 were employed as quality control materials to evaluate the developed method.
Digestion and co-precipitation
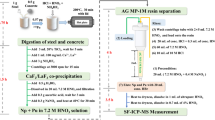
The collected concrete sample was firstly milled to 100 mesh size, and then 0.1–0.5 g aliquot was used for Pu analysis. To obtain the accurate Pu concentration, a total digestion approach was applied to dissolve all compositions (including potential hot particle), as shown in Fig. 1. A volume of 5 mL conc. HNO3, 3 mL HClO4, 2 mL HF and 2 pg 242Pu tracer were added to the PTFE vessel where the concrete sample was placed. After covering the vessel with a cap, the sample was heated at 150 °C for 1 h to allow thorough reaction between the concrete and acids. Then, the cap was removed, leaving the vessel to be heated to dryness at 220 °C. The above evaporation operations were repeated for 1–2 times until no insoluble residue existed after adding 5 mL conc. HNO3. Afterwards, the sample solution together with the subsequent vessel rinse using 5 mL conc. HNO3 were transferred to a 50-mL centrifugation tube. Deionized water was then added to the tube to adjust the sample volume to 35 mL, followed by the addition of 100 mg Ca and 100 mg La. 2 mL 20% TiCl3 was added to convert the valence state of Pu to Pu(III). After adding 7 mL HF, the sample was mixed thoroughly to coprecipitate Pu with CaF2/LaF3. The precipitate was separated from the supernatant by centrifugation at 3000 rpm for 15 min. Finally, the fluoride precipitate was dissolved in 20 mL 3 M HNO3 with the help of 0.5 g H3BO3.

Total digestion and CaF2/LaF3 coprecipitation for concrete samples
Chromatographic separation
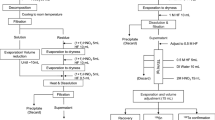
The valence state of Pu was oxidized to Pu(IV) by adding 0.3 g NaNO2 to the prepared sample solution from previous step. Then the sample was loaded onto a TEVA cartridge which had been preconditioned with 10 mL 3 M HNO3. To remove the interfering elements in mass spectrometric determination of Pu isotopes, solutions of 10 mL 3 M HNO3 (tube rinse), 40 mL 1 M HNO3 (remove U, Pb, Tl and Pt), and 10 mL 9 M HCl (remove Th and Bi) were used to rinse the cartridge sequentially. Afterwards, a DGA cartridge (preconditioned by 10 mL 3 M HNO3) was connected to the TEVA cartridge (Fig. 2). The Pu isotopes were subsequently stripped from TEVA cartridge to DGA cartridge by 20 mL 3 M HNO3 − 0.1 M ascorbic acid − 0.02 M Fe(II). Then the TEVA resin was discarded, leaving the DGA resin to be washed by 40 mL 0.1 M HNO3 for further removal of U. Finally, Pu was recovered from DGA cartridge by a rinse of 20 mL 0.5 M HCl − 0.1 M NH2OH·HCl.

Chromatographic separation procedure for concrete samples
ICP-MS measurement
To prepare the sample to a form compatible with ICP-MS measurement, evaporation was firstly conducted for the eluted Pu fraction. Then 4 mL aqua regia was added to decompose NH2OH·HCl and co-eluted organic matter. After heating aqua regia to dryness at 160 °C to dryness, the sample was dissolved in 1 mL 0.88 M HNO3 − 2 pg mL−1 233U. 233U was used here to adjust the sensitivity shift during measurement. In ICP-MS measurement, radioisotopes including 233U (internal standard), 238U (for 238U1H correction [8]), 239Pu (analyte), 240Pu (analyte), 241Pu (analyte) and 242Pu (chemical tracer) were determined in 2 min for each sample.
Results and discussions
Selection of internal standard
Internal standard is usually employed in ICP-MS measurement to adjust the signal intensity change of the analyte resulted from the instrumental sensitivity shift. In literature, isotopes such as 115In, 103Rh, 175Lu and 89Y are commonly used internal standards [11, 13,14,15]. For example, in our previous studies, we used the internal standard 115In in the determination of Pu and Am isotopes [13, 14]. A recent study reported by Narukawa et al. [16] indicated that an ideal internal standard should have close ICP-index value with the analyte, which was defined as a function of atomic mass, 1st ionization potential, particle radii, and number of ionization states. According the calculated ICP-index by Narukawa et al. [16], the ICP-index values for In, Rh, Lu and Y vary a lot from that of Pu. In contrast, the ICP-index values of actinides are similar to each other. Taking into account of this phenomenon, we made the first attempt to use 233U as the internal standard for Pu determination. We compared the signal intensities between 242Pu and 233U which were determined under different parameters, and the results showed that a good correlation was observed (Fig. 3), demonstrating the robustness of using 233U as the internal standard for Pu analysis. Another advantage of this selection was that no 233U was presented in the reagents, preventing the potential interference of impurities in reagents on the signal of internal standard in ICP-MS measurement.

Correlation between 242Pu and 233U signal intensities in ICP-MS measurement
Selection of sample introduction system
In environmental level Pu analysis, high efficiency sample introduction systems (e.g. Apex-Q and Aridus II) were usually employed to increase the instrumental sensitivity of ICP-MS [11, 13]. However, for decommission concrete samples, much higher Pu activity is expected. The increase of instrumental sensitivity is not a key concern. In contrast, the cross contamination in ICP-MS measurement between different samples which is called memory effect should be taken into account. Under this consideration, a scott type spray chamber sample introduction system was used in this study. This system is made of glass and is easy for clearance. The ICP-MS sensitivity of Element XR equipped with a scott type spray chamber is around 5 × 106 cps per ng mL−1 Pu.
Decontamination of interfering elements
In the determination of Pu isotopes by ICP-MS, elements including U, Pb, Bi, Tl, Hg and Pt can cause polyatomic interferences. To assess the decontamination ability of these interfering elements by different extraction resins, we conducted the decontamination experiments for three groups of extraction resins (Table 1). For group 1, only TEVA resin was assessed. While for group 2 and 3, the combinations of TEVA + DGA and TEVA + UTEVA + DGA resins were used. After spiking the 20 mL 3 M HNO3 samples with 10 ng of interfering elements (U, Pb, Bi, Tl, Hg and Pt), the decontamination experiment followed the procedure as described in Fig. 2, except that the DGA procedure was omitted for group 1 samples and that an UTEVA cartridge was added between TEVA and DGA resins during step (4) for group 3 samples. The determined decontamination factors (DFs) for each interfering element were shown in Table 1.
In our previous work [11], we proposed the separation procedure using TEVA + UTEVA + DGA resins to sequentially remove these interfering elements. Considering that the sample size in this study was relatively small (0.1–0.5 g), the requirements on the decontamination ability on interfering elements can be decreased to some extent, especially for the key interfering element U. The elemental concentrations of U in concrete samples ranged from 0.1 to 67.4 μg g−1, as revealed by our investigation for 260 decommission samples. For 0.5 g concrete sample with a U concentration of 67.4 μg g−1, a signal intensity of 1.7 × 1010 cps for 238U was expected in ICP-MS measurement when no decontamination operation was conducted, on a basis of present instrumental sensitivity. To decrease the 238U counting rate to operational blank level (ca. 105 cps), the decontamination factor of U should be higher than 1.7 × 105. As shown in Table 1, the DF (U) of TEVA + DGA resins was 2.3 × 105, higher than the required DF (U). For the decontamination of other interfering elements, it can be learned from Table 1 that the DF values were generally the same for the TEVA + DGA procedure and the TEVA + UTEVA + DGA procedure. Since the latter had been validated to be sufficient for the decontamination of all interfering elements in 2 g solid sample, the TEVA + DGA procedure used in this study was also supposed to be enough for 0.5 g concrete analysis. Thus, we omitted the usage of UTEVA resin and reduced the cost, under the premise of guaranteeing sufficient decontamination ability.
Evaluation of the developed method
Since no standard reference concrete sample is commercially available, we employed the standard reference sediments IAEA-384 and IAEA-385 to assess the accuracy of present method in the determination of Pu. As shown in Table 2, the chemical recoveries of Pu ranged from 72.2 to 89.8% for two investigated materials. The averaged 239+240Pu activity of IAEA-384 was determined to be 105.9 ± 4.9 mBq g−1, consistent with the certified range of 103–110 mBq g−1 [17]. In addition, the averaged 240Pu/239Pu atom ratio was 0.050 ± 0.003, also in good agreement with the certified range: 0.048–0.050. For IAEA-385, similar agreement was observed for 239+240Pu activity and 240Pu/239Pu atom ratio. On a basis of above evaluation, it can be concluded that this method can determine Pu isotopes accurately.
To determine the limit of detection (LOD) of present method, blank concrete samples were analyzed and the results were shown in Table 3. The counting rates of 239Pu and 240Pu in ICP-MS measurement were in the ranges of 3.1–7.9 and 1.0–5.0 cps respectively. Based on the calculated standard deviation of counting rates for blank concrete (Table 3), the concrete sample weight of 0.5 g, Pu chemical recovery of 70% and a ICP-MS sensitivity of 5 × 106 cps per ng mL−1 Pu, the LOD of 239Pu and 240Pu were calculated to be 0.008 mBq g−1 and 0.02 mBq g−1 [14], higher than our previous work due to the lower instrumental sensitivity [11]. However, these LODs were sufficiently enough for the determination of Pu for decommission concrete samples.
Application of the developed method
The whole analytical time of present method takes about 2 days, including 1 day for total digestion and co-precipitation, and 1 day for extraction chromatographic separation and ICP-MS measurement. With the help of 24-hole vacuum box, 22 concrete samples together with a SRM sample and an operational blank sample were analyzed at the same time. This method has been successfully employed to analyze ca. 200 decommission concrete samples by our team, which provided valuable data in the assessment of the decommission facility. The high throughput, short turnaround time, and high chemical recovery of present method allow its potential application in other decommission work where the Pu analysis in concrete sample is also desired.
Conclusions
In this study, a new method was developed to analyze Pu isotopes in decommission concrete. This method firstly used total digestion to dissolve all Pu compositions from 0.1 to 0.5 g concrete sample. CaF2/LaF3 coprecipitation was then followed to remove major matrix. Afterwards, extraction chromatographic separation using TEVA + DGA resins were employed to remove interfering elements. Finally, Pu isotopes were determined by ICP-MS measurement. This method can accomplish the analysis of 22 concrete samples within 2 days, with a relatively high Pu chemical recovery (57–93%). The accuracy of present method in the Pu determination was validated by analyzing two SRMs and the results revealed that both 239+240Pu activity and 240Pu/239Pu atom ratio were accurately determined. In addition, the low LODs (0.008 mBq g−1 for 239Pu and 0.02 mBq g−1 for 240Pu) allow this method to detect low level contamination. Owing to its high throughput, short turnaround time and low LODs, this method had been successfully applied in analyzing ca. 200 decommission concrete samples and more analysis was scheduled in the future.
References
Weinreich R, Bajo S, Eikenberg J, Atchison F (2004) Determination of uranium and plutonium in shielding concrete. J Radioanal Nucl Chem 261:319–325
Xu YH, Qiao JX, Hou XL, Pan SM, Roos P (2014) Determination of plutonium isotopes (238Pu, 239Pu, 240Pu, 241Pu) in environmental samples using radiochemical separation combined with radiometric and mass spectrometric measurements. Talanta 119:590–595
Zheng J, Yamada M, Wang Z, Aono T, Kusakabe M (2004) Determination of plutonium and its isotopic ratio in marine sediment samples using quadrupole ICP-MS with the shield torch system under normal plasma condition. Anal Bioanal Chem 379:532–539
Kim CS, Kim CK, Lee JI, Lee KJ (2000) Rapid determination of Pu isotopes and atom ratios in small amounts of environmental samples by an online sample pretreatment system and isotope dilution high resolution inductively coupled plasma mass spectrometry. J Anal At Spectrom 15:247–255
Muramatsu Y, Uchida S, Tagami K, Yoshida S, Fujikawa TJ (1999) Determination of plutonium concentration and its isotopic ratio in environmental materials by ICP-MS after separation using ion-exchange and extraction chromatography. J Anal At Spectrom 14:859–865
Grate JW, O’Hara MJ, Farawila AF, Douglas M, Haney MM, Petersen SL, Maiti TC, Aardahl CL (2011) Extraction chromatographic methods in the sample preparation sequence for thermal ionization mass-spectrometric analysis of plutonium isotopes. Anal Chem 83:9086–9091
Qiao JX, Hou XL, Roos P, Miró MJ (2010) Rapid and simultaneous determination of neptunium and plutonium isotopes in environmental samples by extraction chromatography using sequential injection analysis and ICP-MS. J Anal At Spectrom 25:1769–1779
Bu WT, Zheng J, Guo QJ, Aono T, Tazoe H, Tagami K, Uchida S, Yamada M (2014) A method of measurement of 239Pu, 240Pu, 241Pu in High U content marine sediments by sector field ICP-MS and its application to Fukushima sediment samples. Environ Sci Technol 48:534–541
Maxwell SL, Culligan BK, Kelsey-Wall A, Shaw PJ (2011) Rapid determination method for determination of actinides in emergency concrete and brick samples. Anal Chim Acta 701:112–118
Qiao JX, Hou XL (2010) Fractionation of plutonium in environmental and bio-shielding concrete samples using dynamic sequential extraction. J Environ Radioact 101:244–249
Wang ZT, Zheng J, Ni YY, Men W, Tagami K, Uchida S (2017) High-Performance method for determination of Pu isotopes in soil and sediment samples by sector field-inductively coupledplasma mass spectrometry. Anal Chem 89:2221–2226
Vajda N, Kim CK (2010) Determination of Pu isotopes by alpha spectrometry: a review of analytical methodology. J Radioanal Nucl Chem 283:203–223
Wang ZT, Zheng J, Cao LG, Tagami K, Uchida S (2016) Method for ultratrace level 241Am determination in large soil samples by sector field-inductively coupled plasma mass spectrometry: with emphasis on the removal of spectral interferences and matrix effect. Anal Chem 88:7387–7394
Wang ZT, Lin JX, Li SX, Guo QJ, Huang WN, Wen W, Dan GP, Tan ZY (2018) Rapid method for accurate determination of actinides (U, Th, Pu and Am) in water samples for emergency response. J Radioanal Nucl Chem. https://doi.org/10.1007/s10967-017-5640-0
Salomon S, Jenne V, Hoenig M (2002) Practical aspects of routine trace element environmental analysis by inductiviely coupled plasma- mass spectrometry. Talanta 57:157–168
Narukawa T, Iwai T, Chiba K (2017) An ICP index for ICP-MS determinations—new selection rules for internal standards in ICP-MS determination and carbon enhancement effect. J Anal At Spectrom. https://doi.org/10.1039/c7ja00132k
Povinec PP, Pham MK (2000) Report on the intercomparison run IAEA-384 radionuclides in fangataufa lagoon sediment. International Atomic Energy Agency, Monaco
Pham MK, Sanchez-Cabeza JA, Povinec PP (2005) Report on the worldwide intercomparison exercise IAEA-385: radionuclides in Irish Sea sediment. International Atomic Energy Agency, Monaco
Acknowledgements
This work was partially supported by the Joint Fund of National Natural Science Foundation of China and Academy of Engineering Physics (Grant No. U1730245) and the National Natural Science Foundation of China (Grant No. 21607139).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, Z., Wen, W., Quan, W. et al. Rapid determination of Pu isotopes for decommission concrete samples by inductively coupled plasma mass spectrometry. J Radioanal Nucl Chem 316, 411–417 (2018). https://doi.org/10.1007/s10967-018-5756-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-018-5756-x