
Abstract
Distribution of Pu(IV) and Pu(V) oxidation states at trace initial concentrations (10−10–10−11 mol L−1) was studied in a liquid- and solid-phase of natural clay and goethite systems. Experiments showed an increase in the concentration of Pu(III) up to 11% at pH 5 in solids of the natural clay −0.1 mol L−1 NaNO3 system containing Pu(IV) after 7-day contact. A kinetic sorption/reduction experiment with goethite suspensions (0.01 mol L−1 NaNO3 containing Pu(V)) indicated the presence of Pu(III) in the solids up to 15%.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Recent developments in ultra-low-level spectroscopy have enabled detailed studies of plutonium concentrations in the environment, and its speciation characterization at ultratrace levels using thermal ionization mass spectrometry (TIMS) and accelerated mass spectrometry (AMS) [1, 2]. Although TIMS and AMS provide accurate and precise determinations of Pu isotopes at low concentrations, these instruments are expensive and their operation requires much experience. From this point of view applications of inductively coupled plasma mass spectrometry (ICP-MS) have certain advantages [3]. These and other techniques [4, 5] have markedly improved the understanding and governing of complex biogeochemical processes of radionuclide migration in the environment providing better prediction of contaminant transport in natural systems. However, there is a lack of information about the radionuclide behavior at low-level concentrations, while sorption parameters are required for modeling to provide better prediction of contaminant transport in natural systems.
Oxidation–reduction reactions are of the top importance in the environmental chemistry. Plutonium redox chemistry is also complicated as well, as speciation strongly affects its environmental behavior [6]. Many previous studies indicated transformation of Pu oxidation state through interactions with surfaces of Mn(IV)-bearing and iron oxide minerals [7–10]. Recent publications have shown that Pu oxidation states can also be strongly affected by natural organic substances [11, 12]. On the basis of lysimetric experiments with well-defined solid sources of Pu(III) (PuCl3), Pu(IV)(Pu(NO3)4 and Pu(C2O4)2, and Pu(VI) (PuO2(NO3)2), it was concluded that Pu(III) may have a wide natural occurrence, especially in acidic environments [13]. Influence of biotic processes on transformation of redox speciation of radionuclides has also received considerable attention [14].
Aqueous actinide species at trace environmental concentrations can be determined exclusively using radiochemical/chemical separations. The procedures for solvent extraction of Pu(IV) at pH 0 and Pu(III), Pu(IV) and Pu(VI) at pH 3–4 using thenoyltrifluoroacetone (TTA) were developed earlier [15]. The TTA extraction procedure was improved [16] when dibenzoylmethane (DBM) was applied to separate actinide elements in the (III), (IV), (V) and (VI) oxidation states in aqueous solutions at neutral pH. The limitations of the TTA extraction procedure were further improved [8] using bis(2-ethylhexyl) hydrogen phosphate (HDEHP), and the extraction conditions necessary to separate Pu(IV), Pu(V) and Pu(VI) in Pu oxidation by manganese oxide studies were well defined [7]. This separation procedure was also applied for investigation of Pu(V) reduction by iron oxides [9].
Recent studies have indicated complicated plutonium behavior in the environment that can be extensively affected both by biotic and abiotic processes. Presence of Pu(III) under oxic and anoxic conditions has been studied, and better understanding of its behavior in the environment has been required. The aim of this work was to study the oxidation state distribution of plutonium at trace concentrations in natural clay and goethite systems.
Experimental
Sample characterisation
Triassic clay from the industrial exploitation site Šaltiškiai in North Lithuania, selected as an engineered barrier for a cement (concrete) based near-surface low and intermediate level radioactive waste repository, and as possible far-field barrier for a high level repository was used in sorption experiments. Selected physical and chemical properties of the Triassic clay are summarized in Table 1. Iron oxides were synthesised using procedures described in literature with some modifications. Details about the synthesis procedures, physical characteristics of the natural clay and goethite, as well as techniques used for mineral characterisation have already been described [17, 18].
Sorption experiments
Sorption was studied under argon atmosphere using 0.01–0.1 mol L−1 NaNO3 solutions. Experiments were based on the standard method for measuring laboratory batch K d values. Pu(IV), Pu(V) and Am(III) were used in sorption experiments at initial concentrations of Pu isotope mixture and 241Am of 1.10 × 10−9–2.50 × 10−11 and 3.20 × 10−11 mol L−1, respectively. Measurements of pH were conducted before and after sorption experiments under continuous Ar flow using WTW SenTix 41 or SenTix 81 pH-electrodes, calibrated with standard buffers DIN 19266 (pH values 4.006, 6.865, 9.180) and a WTW inoLab Multi Level 1 meter. In all experiments ACS reagent grade or higher grade chemicals were used. All solutions were freshly prepared using Milli-Q (Millipore Milli-Q Synthesis A-10) water with TOC < 10 ppb. Dissolved oxygen concentration in solutions was measured using a WTW OxiCal-SL probe and a WTW inoLab Multi Level 1 meter. A possible effect of bacteria was suppressed ultrasonically. Suspensions were shaken for desirable time and the solid-phase was then separated from the liquid-one by centrifugation for 20 min at 10,000–20,000g. The adsorption of radionuclides from solutions to polypropylene bottle walls, determined in the presence of suspended-phase, was found to be negligible (varied from 0.1 to 1%).
Pu and Am in the solution and in the solid-phase were determined after separation using UTEVA and TRU resins (Eichrom Industries), concentrations were then measured by alpha spectrometry or ICP-MS (Finnigan Mat Element 2). A combined standard uncertainty was not higher than 10% at the lowest concentration. 242Pu and 243Am were used as tracers in the separation procedures. Total carbon (TC) and total organic carbon (TOC) in the solids were determined by means of a LECO CS-125. X-ray diffraction (XRD) analyses were conducted using a D8 (Bruker AXS) X-ray diffractometer. A surface area was determined using NOVA 2200 analyzer, and stable iron was measured with a Perkin Elmer Zeeman/3030 AAS-GF.
Oxidation state analysis
Oxidation state distribution analysis employs an ultrafiltration/solvent extraction technique. TTA, HDEHP and DBM were used as described in literature [7–11]. In experiments with Pu(IV) and natural clay a liquid-phase from solids was separated by centrifugation and Pu(III,IV), Pu(IV polymeric), Pu(V) and Pu(VI) were extracted from the aqueous-phase using TTA and HDEHP. Pu was desorbed from the solids with 3 mol L−1 HCl for 1 h. Solutions were filtered using 10 kD centrifugal filter devices (Amicon, Millipore). An aliquot of the filtrate was removed to determine oxidation state distribution by solvent extraction, and fractions of Pu(III,IV), Pu(V), Pu(VI) were separated using TTA and HDEHP as well as Pu(IV) extraction was performed in parallel by TTA from a pH 0 solution. In experiments with Pu(V) and goethite all samples were separated from solids by centrifugation followed by ultrafiltration prior to solvent extraction. Analysis of oxidation state distribution in the solid-phase was carried out using TTA and HDEHP solvent extraction as discussed above. Whereas for analysis of the liquid-phase both TTA/HDEHP and DBM extraction techniques were applied. 241Am(III), 228Th(IV), 237Np(V) and 232U(VI) as oxidation state analogs for Pu(III), (IV), (V) and (VI) as well as 242Pu(IV), 236Pu(III) were used to establish effective leaching conditions of Pu from the solid-phase, and to validate oxidation state analyses procedure.
Results and discussions
Sorption and desorption tests using Am(III), Np(V) and U(VI) as oxidation state analogs for Pu(III), (V) and (VI) were conducted to determine appropriate condition to remove Pu from the solid-phase. Results indicated that Pu can be quantitatively desorbed (up to 96 ± 6%) from solids, in good agreement with data obtained in different laboratories using Pu oxidation state analogs and sequential extraction procedures [7, 10, 17, 18]. Additional experiments performed using 242Pu(IV), 236Pu(III) indicated that Pu(III) and Pu(IV) remain stable during the desorption procedure using 3 mol L−1 HCl.
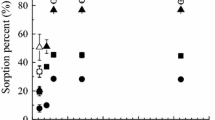
Pu(IV) oxidation state distribution in the system of natural clay (0.1 mol L−1 NaNO3 after 7-day contact time) is shown in Fig. 1. The system is characterised as carbonate-free (TOC < 10 ppb), low oxygen (<50µ mol L−1), and low organic content since only 0.034% of TOC was found in the solids (identification of origin of organic substances is in progress). In addition, Fe(II)- and Fe(III)-bearing minerals were detected in the clay by Mössbauer spectroscopy and XRD analysis. It can be seen that comparatively high content (up to 75%) of Pu(III,IV) was observed at pH 4.86–4.98 while polymeric species of Pu(IV) were found to be the dominant ones at pH 5.76–8.21 in the liquid-phase. The formation of Pu(OH)3+ and Pu(OH) 04 species may explain this distribution [19]. Polymeric species of Pu(IV) can be separated prior to analysis by ultrafiltration, however, such removal can alter the oxidation state distribution of the dissolved plutonium. Thus, polymeric Pu(IV) species were not removed by ultrafiltration, since it was supposed that they are essential for understanding of the whole system and the behaviour of Pu(IV) in natural systems. The solvent extraction techniques used in these experiments help to avoid errors due to Pu redistribution during the solvent extraction procedure. There was very little opportunity to form pseudocolloids in the system since fine particles were removed at the stage of equilibration of solids with the background electrolyte. However, formation of mixed polymeric species of iron and plutonium could affect the results. Analyses of Pu oxidation state determination in the solid-phase were performed after the desorption step using 3 mol L−1 HCl, and Pu(III,IV), Pu(V) and Pu(VI) as well as Pu(IV) at pH = 0 were analysed in parallel. The content of Pu(III) was calculated from balance. Pu(III) was not found in the system at pH (5.76–8.2) while 4.9% and 10.7% of Pu(III) were found at pH 4.86 ± 0.11 and 4.98 ± 0.08, respectively. Iron-bearing minerals such as montmorillonite, siderite, goethite and hematite determined in the studied clay to a great extent account for 10.7% of Pu(III) in solids, because these minerals posses a reductive capacity and can cause the abiotic reductive transformation of plutonium [20]. It should be noted that adsorption experiments with clay and 0.01 mol L−1 NaNO3, where oxidation state distribution analyses were performed using the DBM extraction procedure, also evidenced the presence of Pu(III) in the system. The observed high stability of Pu(IV) in the system may be attributed to two factors: a rapid adsorption to solids, and a strong hydrolysis at near-neutral and high pH.

Dependence of Pu oxidation state distribution on pH in the liquid-phase (L), the solid-phase (S) and the residue (R) of natural clay −0.1 mol L−1 NaNO3 system, [Clay] = 64.2 m2/L, initial [Pu] = 1.10 × 10−10 mol L−1
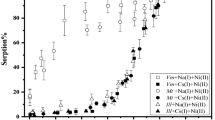
Sorption behaviour of plutonium in natural clay systems can be affected by a variety of reactions, including surfaces complexation, oxidation and reduction. Since iron oxides and minerals found in Triassic clay coatings (Table 1) could alter the plutonium redox speciation in a different way, experiments were conducted with pure phases, and as the first approach with synthetic goethite and 0.01 mol L−1 NaNO3 (at pH = 4.89–5.01). The oxidation state transformation of Pu(V) over time in the liquid- and solid-phase for the system containing Pu(V) are presented in Fig. 2. The obtained data revealed the complicated oxidation–reduction behaviour of Pu(V) in the system. It seems that Pu(V) after adsorption was reduced to Pu(IV), while a small portion reduced to Pu(III) was later oxidised to Pu(IV). Furthermore, higher amount of Pu(III) was found in the solids (up to 15%) in comparison with the liquid-phase, where it varied from 1 to 3%. It should be noted that the reduction of Pu(IV) to Pu(III) in the natural clay −0.1 mol L−1 NaNO3 system was expected because of rather high content of siderite determined in the studied clay coatings (Table 1). Probably a long contact time and oxidation of Pu(III) in the system resulted in the low concentrations of Pu(III) measured in the solid-phase. However, Pu(III) concentrations found in the goethite solids after sorption are rather surprising and require additional studies, whereas minor concentrations of Pu(III) in the aqueous-phase are probably due to the absence of any ligand, capable to stabilise it in the system. We suppose that Pu(III) was quantitatively desorbed from the goethite solid since the amount of 241Am found in the residue after the desorption step did not exceed 5%, however, the Pu amount in the residue has been increasing with contact time (Fig. 3). This strong bonding to the solids can be explained by the Pu sorption to crystalline iron oxides present in the clay coatings, which was studied in kinetic sorption experiments followed by sequential extraction and Mössbauer spectroscopy [18]. The slightly different adsorption/reduction patterns observed in this work when compared with tests performed by other researchers [10] are perhaps due to different experimental conditions and Pu concentrations used in experiments.

Pu oxidation state distribution in the liquid-phase (L), the solid-phase (S) and the residue (R) of synthetic goethite −0.01 mol L−1 NaNO3 system, [α-FeOOH] = 172 m2/L, pH 4.89–5.01, initial [Pu] = 2.50 × 10−11 mol L−1

Content of Pu and Am in the goethite residue after desorption
Conclusions
The oxidation state distribution at trace initial Pu concentrations in liquid- and solid-phase of the natural clay and goethite systems was studied. Existing solvent extraction techniques were verified with respect to determination of Pu(III) in the liquid- and solid-phase. Pu(III) was desorbed quantitatively from the solids, no oxidation of Pu(III) and Pu(IV) was observed during desorption procedure. The reduction of Pu(IV) by natural clay containing iron-bearing minerals in the pH range from 4.22 to 4.98 was found, however, no reduction was detected at the near-neutral and high pH. In kinetic sorption experiment with goethite −0.01 mol L−1 NaNO3 suspensions, Pu(V) was mainly reduced to Pu(IV), and a small portion of Pu(III) found at the beginning in the solids (up to 15%) was later oxidised to Pu(IV).
References
Kurosaki H, Chang D, Inn KGW (2006) J Radioanal Nucl Chem 269:279
Povinec PP (2005) J Radioanal Nucl Chem 263:413
Povinec PP, La Rosa J, Lee S-H, Muslow S, Osvath I, Wyse E (2001) J Radioanal Nucl Chem 248:713
La Rosa J, Gastaud J, Lagan L, Lee S-H, Levy-Palomo I, Povinec PP, Wyse E (2005) J Radioanal Nucl Chem 263:427
Lee S-H, La Rosa J, Gastaud J, Povinec PP (2005) J Radioanal Nucl Chem 263:419
Choppin GR (2000) Radiochim Acta 91:3645
Morgenstern A, Choppin GR (2002) Radiochim Acta 90:69
New MP, Hoffman DC, Roberts KE, Nitsche H, Silva RJ (1994) Radiochim Acta 66:265
Powell BA, Fjeld RA, Kaplan DI, Coates JT, Serkiz SM (2004) Environ Sci Technol 38:6016
Powell BA, Fjeld RA, Kaplan DI, Coates JT, Serkiz SM (2005) Environ Sci Technol 39:2107
Andre C, Choppin GR (2000) Radiochim Acta 88:613
Banik NL, Buda RA, Bürger S, Kratz JV, Trautmann N (2007) J Alloys Compd 444–445:522
Kaplan DI, Powell BA, Duff C, Demikanli DI, Denham M, Fjeld A, Molz FJ (2007) Environ Sci Technol 41:7417
Panak PJ, Nitsche H (2001) Radiochim Acta 89:499
Foti SC, Freiling EC (1964) Talanta 11:385
Saito A, Choppin GR (1983) Anal Chem 55:2454
Lujanienė G, Šapolaitė J, Amulevičius A, Mažeika K, Motiejūnas S (2006) Czechoslov J Phys 56:103
Lujanienė G, Motiejūnas S, Šapolaitė J (2007) J Radioanal Nucl Chem 274:345
Knopp R, Neck V, Kim JI (1999) Radiochim Acta 86:101
Lee W, Batchelor B (2003) Environ Sci Technol 37:535
Acknowledgements
The research was supported by Ministry of Education and Science of the Republic of Lithuania and the Lithuanian State Science and Studies Foundation projects V-19/2009, NKS-B SPECIATION project (2008–2009), FP7 RECOSY, grant No 212287.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lujanienė, G., Šapolaitė, J., Radžiūtė, E. et al. Plutonium oxidation state distribution in natural clay and goethite. J Radioanal Nucl Chem 282, 793–797 (2009). https://doi.org/10.1007/s10967-009-0175-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10967-009-0175-7