
Abstract
Starch is an abundant natural and renewable carbohydrate polymer, with cation sorption deficiency due to its high crystallinity and amylose content which causes substantive mass transfer resistance during adsorption. Therefore, the present study synthesized polyacrylonitrile grafted starch (graft-starch) that was subjected to heat treatment and saponification reaction to facilitate a partial transformation of the available amide groups (due to copolymer grafting) into carboxylate groups. The reactions afforded the desired polyacrylonitrile-grafted starch hydrogel (‘Poly-g-Hyd’) which was further utilized for zinc ion uptake. It was observed that the polar nitrile (CN) groups of the grafted polyacrylonitrile substantially improved the ‘Poly-g-Hyd’ water absorption capacity and cation binding interaction, as well as entrenched stable network chains that are insoluble in water. The water absorption and zinc ion sorption capacities of 490 g water/g ‘Poly-g-Hyd’ and 508.85 mg/g, were respectively recorded. Furthermore, the optimum adsorption conditions were established at pH 5.0, 20 min and 0.3 g/L, while the Freundlich and pseudo-second-order models provided the best fit to the isotherm and kinetic data, respectively. The study, therefore, demonstrated the effectiveness of ‘Poly-g-Hyd’ in the uptake of aqueous zinc ions.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The rapid industrialization and world population growth have caused a surge in ecosystem toxicity due to the presence of aqueous heavy metal contamination [1,2,3]. The ecotoxicological dangers associated with these heavy metal ion accumulations in the aquatic environment is primarily linked to their poor degradability [4]. Our choice adsorbate, zinc ions (Zn 2+) has extensive application in process industries (in steelwork, galvanization, brass plating, fibre manufacturing, dye & pigment, etc.) [5]. When the contaminated waste streams from these industries are poorly treated before discharge into the environment, zinc ions could find their way into the food chain of living species [6]. Although zinc is an essential element, its high-level accumulation in living species could cause grave health damage [7, 8]. Some of its adverse health impacts include depression, inactivity, abnormal neurological functionality, etc. [9]. The World Health Organization (WHO) stipulated a 5.0 mg L− 1 limit for aqueous zinc ion contamination [10], however, literature reports showed a zinc ion concentration in the range of 1 ≥ Zn2+ ≥ 48000 mgL−1 from different waste streams [4].
Considering the aforementioned health impacts of heavy metal ion pollution, worthwhile research into the various approaches for the reduction of their (heavy metal ions) concentration in waste streams to within the WHO permissible limits is plausible. Various approaches, such as coag-flocculation, membrane filtration, ion exchange, etc. have been previously adopted for aqueous zinc ion treatment, with several associated drawbacks (like high cost, poor treatment efficiency, etc.) [8]. Meanwhile, biosorption has been regarded as a promising treatment option due to its treatment efficiency, cost-effectiveness, operational flexibility, etc. [11,12,13].
Many adsorbents, including natural carbohydrate polymers (starch), have been extensively reported as efficient for aqueous heavy metal ion biosorption. These materials were either used raw [14,15,16] or with some form of chemical modifications [17, 18]. Meanwhile, the cation sorption capacity of these raw starch is limited by their high crystallinity and amylose content [19]. They also hamper the diffusion of the cations into the polymer chelation sites [5]. To forestall such limitation, researchers have attempted the chemical modification/functionalization (via improvement of the available functional groups) of these native carbohydrate polymers, aimed at yielding efficient adsorbents, with superior heavy metal sorption capacity [8]. This is because their (carbohydrate polymers) cation sorption efficiency is a function of the electrostatic complexation, governed by the adsorbate-adsorbent surface charge differences [20]. Polymer sorbent functionalization could be achieved through different chemical techniques like copolymer grafting, hydrolysis, etc. [21]. Copolymer grafting (using suitable monomer), an important functionalization avenue could successfully introduce important metal-binding sites such as carboxyl, amino group, etc., as well as significantly modify the starch/polymer network [5, 22].
Therefore, the present study involved the synthesis of polyacrylonitrile grafted starch (graft-starch) which was subjected to heat treatment and subsequent saponification reaction to facilitate a partial transformation of the entrenched amide groups into carboxylic groups. The reactions afforded the desired polyacrylonitrile-grafted starch hydrogel (‘Poly-g-Hyd’) which was then utilized for zinc ion uptake. The objectives of the study are stated thus; (a) to measure the nitrogen content of the synthesized hydrogel, as well as their swelling capacity (b) to characterize the synthesized ‘Poly-g-Hyd’ using FTIR, SEM, and SEM–EDX, (c) to evaluate the effect of contact time, adsorbent dosage and adsorbate solution pH on the adsorption capacity (c) to elucidate the isotherm, kinetics and mechanism of the adsorption process.
Materials and methods
Materials
The starch precursor (maize starch) used in the study was obtained from the Egyptian Starch and Glucose Manufacturing Company, Cairo, Egypt. The acrylonitrile, ammonium cerium (IV) nitrate (CAN), zinc acetate, caustic soda, hydrogen trioxonitrate (v) acid, ethanoic acid, acetone, methanol, ethyl alcohol and all other employed reagents were all laboratory grade chemicals, supplied by Merck, Germany.
Methods
Synthesis of the grafted starch
The copolymerized (graft) starch was synthesized following the procedure earlier reported by Abdel-Halim [23], with slight modifications. The procedure involved the gelatination of a specific amount of maize starch in suitable (enough to form a gel) volume of water followed by heat treatment of the resultant starch gel (at 85 °C for 30 min) and subsequent addition (30 ± °C and stirring at 150 rpm ) of a specific volume of acrylonitrile, together with a binary mixture of freshly prepared ammonium cerium (iv) nitrate and 1 N HNO3 to afford the starch copolymer. The reaction by-products (homopolymers) were further eliminated via the precipitation of the starch copolymer in N, N-dimethylformamide (DMF). The resultant precipitates (homopolymer-free graft-starch) was obtained, washed with deionized water and then oven-dried at 50 °C to constant dry weight.
Following the micro-Kjeldahl method [24], the % nitrogen content of the graft-starch was estimated, while the % graft yield was evaluated from Eq. (1) [17, 25].
NB
53 and 14 is the molar mass of acrylonitrile and Nitrogen (g/mol), respectively.
Hydrogel synthesis
10 mL of 0.7 N sodium hydroxide solution was added to 1.0 g of the graft-starch contained in a loosely stoppered (to permit ammonia evolution) flask, followed by mechanical stirring at 150 rpm and 95 °C. A subsequent change in the colour of the solution (from deep red to light yellow informs the climax of the saponification reaction. Furthermore, the saponified starch was contacted with a predetermined volume of methanol, stirred for 5 min and excess volume of ethanol was subsequently added to the mixture. The precipitate so obtained was washed with acidified ethanol (until neutral pH), filtered off, oven-dried (60 °C, 3 h) to afford the superabsorbent hydrogels (‘Poly-g-Hyd’).
Hydrogel swelling capacity measurement
The measurement was achieved via the impregnation of a predetermined amount of the synthesized ‘Poly-g-Hyd’ in a suitable volume of distilled water (at 30 ± 3 °C ) until its saturation. The fully swollen ‘Poly-g-Hyd’ was recovered as filtrate, drained and reweighed. Its water absorption capacity was then evaluated from Eq. (2) [23, 25].
Where M1 and M2 are the Masses of swollen ‘Poly-g-Hyd’ and dry ‘Poly-g-Hyd’.
Hydrogel characterization
The surface chemistry/functional groups on the raw and spent hydrogel, as well as that of the hydrogel precursor, was elucidated via FTIR (Perkin–Elmer Spectrum1000 spectrophotometer) at 4000–400 cm− 1 and scan-interval of 1 cm− 1 (over 120 scans). Similarly, the surface morphology was obtained via SEM measurement [TESCAN CE VEGA 3 SBU (117-0195- Czech Republic)] at 1000x magnification. Before the SEM analysis, the samples were mounted on a standard microscope stub and coated with a thin gold layer. Consequently, the presence of the adsorbed zinc ions on the adsorbent was informed from the Energy-dispersive X-ray (EDX) patterns.
Adsorption experiments
By dissolving an appropriate amount of zinc acetate in deionized water, the zinc ion stock solution (1 g L− 1) was obtained. Meanwhile, the working zinc ion concentrations were further achieved from the serial dilution (using deionized water) of the stock solution.
During the adsorption experiment, a predetermined amount of ‘Poly-g-Hyd’ was charged into the adsorbate solution (100 mL) contained in a 250 mL conical flask. The mixture was stirred mechanically on a magnetic stirrer (Stuart, Model UC151/120V/60) at 150 rpm and 30 °C. At a specified interval, test samples were withdrawn from the mixture, filtered through Whatman No. 41 filter paper and the remaining Zn (II) ion concentration was measured using atomic absorption spectrophotometer (AAS, ZA3000, HITACHI, Japan). By varying the solution pH using either 0.1 M HNO3 or 0.1 M NaOH as appropriate, the effect of pH studies was investigated in the pH range of 2.0–7.0. Similarly, the effect of time (0–60 min) and adsorbent dosage (0.3-4.0 g/L) was also investigated.
The associated adsorption parameters such as the % uptake efficiency (RE %), as well as the amount of zinc ion adsorbed at equilibrium (qe) are calculated from Eqs. (3) and (4), respectively.
Where Co = Initial zinc ion concentration (mg L− 1), Ce = zinc ion concentration at equilibrium (mg L− 1), W = Mass of ‘Poly-g-Hyd’ used (g), V = volume of adsorbate solution used (L).
Results and discussion
Adsorbent characterization result
Physicochemical analyses
The % nitrogen content on the synthesized hydrogel (‘Poly-g-Hyd’) as shown in Table 1 provide information on the extent of the copolymer grafting. The result shows a depletion in the % nitrogen content of the grafted starch following the saponification reaction towards the hydrogel synthesis. This finding could be linked to the transformation of some of the cyano groups on the grafted starch to amide (-CONH2) and carboxylate (-COONa) groups, although the carboxylate groups were subsequently at the later stage of the hydrogel synthesis process.
According to the value depicted in Table 1, ‘Poly-g-Hyd’ showed a relatively high absorption capacity of 490 g water/g ‘Poly-g-Hyd’, which could be due to the structural changes occurring during the sequence of steps involved in the copolymer grafting. For instance, it is believed that the gelatination effect could be responsible for significant physicochemical changes (such as molecule disorientation, free volume magnification, etc.) observed on the starch structure [17]. Similarly, the polymer crosslinking due to the copolymer grafting significantly improved the ‘Poly-g-Hyd’ structural network [23].
Scanning Electron micrograph
The morphology of the maize starch (precursors), ‘Poly-g-Hyd’ and Zn (II)-loaded ‘Poly-g-Hyd’ was obtained using SEM imagery and presented in Fig. 1. As shown in Fig. 1a, the raw starch resembled a woollen matrix, with fibrous interconnections and substantial surface heterogeneity. Meanwhile, the hydrolysed grafted starch (Fig. 1b) showed better surface aggregated patches, with evidence of particle agglomeration into more pronounced irregular shapes of superior surface roughness and porosity. Furthermore, the previously intact starch polymeric networks observed in Fig. 1a partially crumbled into loosely bound biofilm-like amorphous matrices (Fig. 1b) sequel to hydrogel formation. This improved surface roughness and porosity is a direct consequence of the precipitation stage, which eliminated the solubilized low molecular weight homopolymer. Furthermore, the flake-like smoothened surface morphology, with no surface roughness and porosity observed in Fig. 1c, is an indication of the presence of the adsorbed heavy metal cation on the adsorbent pores.

SEM images for (a) maize starch (b) ‘Poly-g-Hyd’ and (c) Zn (II)-loaded ‘Poly-g-Hyd’
Energy dispersion X-ray (EDX) study
Elemental mapping and % mass distribution on the spent adsorbent (Zn (II)-loaded ‘Poly-g-Hyd’) is inscribed into Fig. 2. The considerable presence of carbon (13.50 %) and oxygen (21.45 %), which are the main component of starch compounds was observed in the EDX mapping spectrum, as well as the presence of the adsorbed zinc (11.59 %) ion. The observed presence of zinc ions in the post adsorption EDX spectrum confirms their adhesion onto the adsorbent. Furthermore, the nitrogen group that was introduced onto the starch material during its copolymerization with the acrylonitrile was not identified in Fig. 2. This development confirmed the exhaustion of these nitrogen groups as a result of cation chelation.

Energy dispersion x-ray mapping spectrum
Surface chemistry
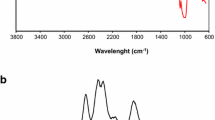
Figure 3 shows the FTIR spectra for raw starch, grafted starch, ‘Poly-g-Hyd’ and Zn (II)-loaded ‘Poly-g-Hyd’. The H-bonded OH stretch (assigned to bands existing around 3400 cm− 1), C - H stretch (around 2900 cm− 1), amide/carboxylate C = O stretch (1720–1550 cm− 1), C – C stretch (1400–1200 cm− 1) and C – O stretch (around 1150 cm− 1) are the major functional groups common in all the spectra (Fig. 3). Upon the comparison of the different spectra, it was observed that the C - H stretch of the raw starch was suppressed sequel to copolymer grafting operation. Similarly, the C ≡ N and C ≡ C groups, respectively identified on the grafted starch and raw starch spectra consequently disappeared in the ‘Poly-g-Hyd’ (Fig. 3). Furthermore, the diminished C = O stretch peak on the grafted and raw starch became more pronounced on the ‘Poly-g-Hyd’ spectra. A conspicuous nitrile (C ≡ N) group identified at 2237 cm−1 on the grafted starch spectra affirmed the success of the copolymer grafting. The FTIR spectra of the post adsorption sample did not differ much from that of the unused adsorbent, except for the observed reduction in peak intensity.

FTIR spectra for (a) raw starch (b) grafted starch (c) ‘Poly-g-Hyd’ (d) Loaded ‘Poly-g-Hyd’
Effect of the process variables
Effect of solution pH
The pH of the adsorbate solution and the surface charge of the adsorbent (point of zero charge) is crucial for determining the extent of the adsorbate-adsorbent complexation and interaction [26]. Considering the pHpzc of ‘Poly-g-Hyd’, which was recorded at pH 2.4 (plot not shown), the operational pH for the study was obtained at pH above pH 2.4 (Fig. 4a). This observation is so much as expected because, at a lower pH (pH < pHPZC), the adsorption sites on the ‘Poly-g-Hyd’ were mostly protonated, such that the hydrogen ion (H+) tends to compete with cations. A steady increment in the solution pH towards and above the pHPZC expectedly resulted in substantive negativity of the adsorbents’ surface charge, thus allowing for increased cation affinity and subsequent binding [27]. For instance, no observable zinc cation uptake occurred at pH 2.0, hence the adsorption capacity at that pH was 0. Similarly, only 10 % (36.54 mg/g) of the total adsorption capacity recorded at optimum condition was realised at pH 3.0. Meanwhile, at an optimum condition of pH 5.0, the highest adsorption capacity of 365.44 mg/g was recorded.
Effect of adsorbent dosage
The effect of the adsorbent dosage on cation uptake capacity and removal efficiency was simultaneously investigated and the finding is shown in form of a plot in Fig. 4b. It is observed that the removal efficiency varies inversely with the adsorption capacity. Meanwhile, the relationship between that adsorbent dosage and adsorption capacity will be emphasized in this discussion. This is because the adsorption capacity is a function of the intrinsic adequacy of the adsorbent, while the initial adsorbate concentration significantly affects the removal efficiency [28].
Analysis of Fig. 4b showed that an increase in the adsorbent mass (from 0.3 to 1.0 g/L) resulted in a sharp decline (about 47 % reduction) in the ‘Poly-g-Hyd’ adsorption capacity. This trend of sustained decrease in the adsorption capacity persisted with an increase in the amount of ‘Poly-g-Hyd’ at every instant. Hence, the adsorption capacity ultimately reduced from 368.761 mg/g (at 0.3 g of ‘Poly-g-Hyd’/litre of adsorbate) to as low as 85.64 mg/g (at 4.0 g of ‘Poly-g-Hyd’/litre of adsorbate). This observation could be linked to the possible active site clogging, overcrowding and their subsequent agglomeration at high adsorbent dosage [26, 29]. Therefore, an optimum ‘Poly-g-Hyd’ mass of 0.3 g/L was subsequently adopted in the study.
Effect of contact time and initial concentration
Adsorption is majorly a mass transfer operation; thus, a sustained extension of adsorption duration is expected to improve the rate of adsorbates’ molecular diffusion from the bulk liquid phase, through the solid-liquid interface and into the adsorbents active sites. The effect of contact time at varying adsorbate initial concentrations was investigated and the finding presented in Fig. 4c. The results showed that about 91 % (384.125 mg/g) and 93 % (328.153 mg/g) of the optimum adsorption capacities, at the initial concentration of 500 mg/L and 400 mg/L, respectively was realized within the first 5 min of adsorption operation. This observation speaks volumes of the affinity between the ‘Poly-g-Hyd’ and the zinc cation, due to the recorded fast adsorption kinetics [11]. Meanwhile, the remaining 9 % and 7 % increment in the optimum adsorption capacity for 500 mg/L and 400 mg/L systems, respectively occurred at the brink of adsorption equilibration at 20 min contact time. This finding suggests a possible saturation of the ‘Poly-g-Hyd’ active sites [30]. The rapid adsorption rate observed within the first 5 min of operation is related to the availability of a large number of zinc cations (which increased with an increase in adsorbate concentration) and ‘Poly-g-Hyd’ active sites. Conversely, the abated adsorption rate experienced as the process proceeded beyond 5 min until the attainment of equilibrium at 20 min resulted from the active site saturation and reduction in the adsorbate concentration at the bulk liquid phase [31]. Further observation of Fig. 4c shows that the adsorption capacity increased as the initial adsorbate concentration. This phenomenon characterizes a surface reaction, where the concentration induced driving force between the zinc cations and the ‘Poly-g-Hyd’ active sites is strengthened [32, 33].

The plot of the effect of (a) solution pH on the adsorbed amount (b) adsorbent dose on the % removal and adsorbed amount (c) variation of contact time and initial concentration on the adsorbed amount
Equilibrium studies
In this study, the experimental equilibrium data were modelled with four (4) isotherm models namely Langmuir, Freundlich, Temkin, and Dubinin–Radushkevich (D-R) models [34] to obtain the model of best fit. However, the smaller the average percentage error (APE) for a given model as evaluated from Eq. (5), the better the model fit due to reduced error variance between the experimental and model-predicted adsorption capacity values. The import of the respective isotherm model parameters to the present adsorption system is discussed in the subsequent subsections, while the combined isotherm plot and parameter table for all the models are presented in Fig. 5; Table 2, respectively.
Where ‘n’ is the number of the experimental data point.
Langmuir model
This model is hinged on the fundamental assumption of monolayer adsorption onto a homogenous adsorbent surface, with identical/energetically uniform adsorption sites. The nonlinear Langmuir model equation and the associated favorability constant (RL) is expressed as Eqs. (6)–(7) [35]. Where RL > 1, RL = 1, RL = 0 and 0 < RL < 1 implies unfavourable, linear, irreversible and favourable nature of the adsorption process, respectively.
According to Table 2, the model showed a sufficiently large maximum adsorption capacity of 508.859 mg/g. Similarly, the models’ separation factor, RL depicted a positive value of 0.016 (0 < RL < 1), thus suggesting adsorption favorability. Its coefficient of determination (R2) and APE values of 0.937 and 4.86, respectively is relatively good and within a suitable range but these values are inferior to those recorded by Freundlich and Temkin model. Based on the model of best fit selection criteria, the Langmuir model cannot be regarded as the best fit, however, its predicted data provided a reasonable fit to the experimental data.
Freundlich model
This model is synonymous with heterogeneous multilayer adsorption and uneven adsorption energy distribution [36]. The model is mathematically expressed as Eq. (8), while the model constant (nF) value suggests the extent of the adsorbent-adsorbate affinity.
The fitting of the equilibrium experimental data to this model (Fig. 5) depicted the highest R2 and the lowest APE values of 0.987 and 2.80, respectively (Table 2). Based on these values, the model predicted data is believed to have provided the best description of experimental data. Similarly, the evaluated nF value of 3.05 is well within the expected range (1 < nF < 10), thus implying a favourable interaction between the zinc cation and the ‘Poly-g-Hyd’ adsorbent. Consequently, the zinc cation sustained a multilayer binding onto the ‘Poly-g-Hyd’ surface.
Temkin model
The adsorbate-adsorbent interaction during adsorption is correlated by the Temkin model [37]. The model postulates a uniform distribution of adsorption energy, with a direct variation between the heat of adsorption and surface coverage. Its nonlinear equation is expressed as Eq. (9).
The model parameters and their associated goodness of fit (R2 and APE) values that are presented in Table 2 are reasonable good. For instance, its R2 value of 0.917 implies that about 90 % of the experimental data was explained by the model predicted data. Similarly, the bT constant which is related to the adsorption energy variation depicted a positive value. Hence, the increase in temperature of the adsorption system will have a detrimental effect on the entire sorption process. Despite the high R2 and relatively low APE value, the comparison of the model and experimental isotherm curve (Fig. 5) showed that the Temkin model curve varied widely from the experimental equilibrium curves. Therefore, although the Temkin isotherm can describe the zinc cation sorption onto ‘Poly-g-Hyd’, it’s fitting ability still fell short of those provided by the Freundlich model.
Dubinin–Radushkevich (D-R) model
As an empirical model, Dubinin–Radushkevich isotherm [38], effectively relates the mechanism of heterogeneous surface adsorption, with Gaussian distribution of energy [39, 40]. The model term ‘E’ which denote the mean free energy and is evaluated from Eq. (10) offers a distinction between physical (E < 8 kJ/mol) and chemical adsorption [41]. The D-R model equation is shown in Eq. (11).
This model showed a reasonably high R2 value, however, it’s the magnitude of its APE value of 6.29 was the largest of all. Figure 5 also showed a wide variation between the experimental isotherm curve and the model curve. This finding portrays the model’s inefficiency at describing the experimental equilibrium data. Meanwhile, the magnitudes of the D-R mean energy, E (42.25 KJ/mol), as evaluated from Eq. (10), is larger than 8 kJ/mol; a suggestion for the dominance of chemisorption [42, 43].

A plot showing the Experimental Langmuir, Freundlich, Temkin and D-R isotherm data
Kinetic studies
The experimental kinetic data was modelled using two (2) classical and commonly applied models, the pseudo-first-order and pseudo-second-order [44, 45]. The mathematical expression of these models, their respective supporting parameters and the import of the respective model parameters (at different initial adsorbate concentrations) to the present adsorption system are discussed elaborately in the following subsections. The judgement on the goodness of fit of the respective model is made based on the magnitude of the APE- values which are evaluated from Eq. (5).
Pseudo-first-order (PFO) model
The PFO [46] postulates that the adsorption rate constant (k1) varies inversely as the adsorbate concentration (ce), with a characteristic first-order reaction mechanism. The nonlinear form of the PFO model is presented in Eq. (12).
The models’ plot and the corresponding parameters obtained for the 400 and 500 mg/L adsorption system are presented in Fig. 6; Table 3, respectively. Observation from the Table showed that the rate constant (k1) and calculated adsorption capacity (qe cal) increased correspondingly as the initial concentration. Meanwhile, theoretical maximum adsorption capacity (qe, Cal) values of 370.34 mg/g (for 400 mg/L system) and 420.0 mg/g (for 500 mg/L system) are consistent with the experimentally derived uptake amount (qe @ 400 mg/L = 368.76 mg/g and qe @ 500 mg/L = 419.79 mg/g). Furthermore, the coefficient of determination (R2) was in all cases greater than 0.9, while relatively low APE values were also recorded. These results are indications of the good fitting of the PFO model. The experiment-isotherm plot shown in Fig. 6 depicted a close correlation between the experimental curve and the PFO model curve, an indication for the PFO model prediction adequacy.
Pseudo-second-order (PSO) model
The PSO [47] model assumes that the adsorption rate is a direct function of the available adsorption sites on the adsorbent and also provides information on the adsorbents’ adsorption capacity. The PSO nonlinear equation is expressed as Eq. (13).
The models’ plot and the corresponding parameters obtained for the 400 and 500 mg/L adsorption system are presented in Fig. 6; Table 3, respectively. In comparison with the PFO data, the PSO model depicted lower APE values and higher R2-values; thus, it provides a better fit to the experimental data. Just like in the case of the PFO model, its adsorption capacity increased as the initial adsorbate concentration, however, the k2-value remained constant for both adsorption systems (Table 3). This signifies that the PSO rate constant (k2) is independent of bulk liquid phase metal ion concentration, with increased adsorbate-adsorbent affinity as the initial concentration increased. The experiment-isotherm plot shown in Fig. 6 depicted a closer correlation between the experimental curve and the PSO model curve when compared with that of the PFO model. Therefore, it is believed that the PSO model provided the best fit to the experimental kinetic data.

Plots of the Experimental, Pseudo-first-order, and Pseudo-second-order kinetics data at varying initial concentrations
Conclusions
Hydrogel based adsorbent (‘Poly-g-Hyd’) was successfully synthesized via the saponification of acrylonitrile grafted starch precursor. The extent of the initial copolymer grafting was determined from the % nitrogen content of the graft product. Meanwhile, due to the transformation of some of the cyano groups on the grafted starch to amide (-CONH2) and carboxylate (-COONa) groups, the synthesized hydrogel (‘Poly-g-Hyd’) depicted a lower % nitrogen content when compared to that of the grafted starch. Furthermore, the ‘Poly-g-Hyd’ showed a relatively high-water absorption capacity of 490 g water/g ‘Poly-g-Hyd’, which could be due to the structural changes occurring during the sequential stages of gelatination and crosslinking involved in the adsorbent synthesis protocol. The maximum zinc ion uptake was recorded at pH 5.0, 20 min and 0.3 g/L adsorbent concentrations. The adsorption experimental data was effectively modelled using equilibrium and kinetic models, while the effect of some process variables on the ‘Poly-g-Hyd’ adsorption capacity was also elucidated in the study.
Data availability statement
The raw/processed data required to reproduce these findings cannot be shared at this time as the data also forms part of an ongoing study.
References
Hashem A, Aniagor CO, Badawy SM, Taha GM (2021) Novel application of the esterification product of 2, 3-dihydroxybutanedioic acid and cellulosic biomass for cobalt ion adsorption. Korean J Chem Eng. https://doi.org/10.1007/s11814-021-0872-7
Hashem A, Al-Anwar A, Nagy NM, Hussein DM, Eisa S (2016) Isotherms and kinetic studies on adsorption of Hg (II) ions onto Ziziphus spina-christi L. from aqueous solutions. Green Process Synth 5(2):213–224
Hashem A, Badawy SM, Farag S, Mohamed LA, Fletcher AJ, Taha GM (2020) Non-linear adsorption characteristics of modified pine wood sawdust optimised for adsorption of Cd (II) from aqueous systems. J Environ Chem Eng p. 103966
Deliyanni E, Peleka E, Matis K (2007) Removal of zinc ion from water by sorption onto iron-based nanoadsorbent. J Hazard Mater 141(1):176–184
Abdel-Halim E, Al-Deyab SS (2014) Preparation of poly (acrylic acid)/starch hydrogel and its application for cadmium ion removal from aqueous solutions. React Funct Polym 75:1–8
Jain C (1996) Application of chemical mass balance to upstream/downstream river monitoring data. J Hydrol 182(1–4):105–115
Hashem A, Abdel-Lateff A, Farag S, Hussein D (2008) Treatment of alhagi residues with tartaric acid for the removal of Zn (II) ions from aqueous solution. Adsorp Sci Technol 26(9):661–678
Chakraborty R, Asthana A, Singh AK, Jain B, Susan AB (2020) Adsorption of heavy metal ions by various low-cost adsorbents: a review. Int J Environ Anal Chem p. 1–38
Srivastava VC, Mall ID, Mishra IM (2008) Removal of cadmium (II) and zinc (II) metal ions from binary aqueous solution by rice husk ash. Colloids Surf A Physicochem Eng Asp 312(2–3):172–184
Hawari A, Rawajfih Z, Nsour N (2009) Equilibrium and thermodynamic analysis of zinc ions adsorption by olive oil mill solid residues. J Hazard Mater 168(2–3):1284–1289
Aniagor CO, Elshkankery M, Fletcher A, Morsy OM, Abdel-Halim E, Hashem A (2021) Equilibrium and Kinetic Modelling of Aqueous Cadmium Ion and Activated Carbon Adsorption System. Water Conserv Sci Eng 6:95–104
Mohamed LA, Aniagor CO, Hashem A (2021) Isotherms and kinetic modelling of mycoremediation of hexavalent chromium contaminated wastewater. Cleaner Engineering Technology 4:100192
Wang J, Chen C (2009) Biosorbents for heavy metals removal and their future. Biotechnol Adv 27(2):195–226
Božić D, Gorgievski M, Stanković V, Štrbac N, Šerbula S, Petrović N (2013) Adsorption of heavy metal ions by beech sawdust–Kinetics, mechanism and equilibrium of the process. Ecol Eng 58:202–206
Mohammed RR (2012) Removal of heavy metals from waste water using black teawaste. Arab J Sci Eng 37(6):1505–1520
Hashem A, Aniagor C, Taha G, Fikry M, Utilization of Low-cost Sugarcane Waste for the Adsorption of Aqueous Pb(II): Kinetics and Isotherm Studies. Current Research in Green Sustainable Chemistry, 2021. 4, 100056, https://doi.org/10.1016/j.crgsc.2021.100056
Hashem A, Fletcher A, El-Sakhawy M, Mohamed LA, Farag S (2020) Aminated Hydroximoyl Camelthorn Residues as a Novel Adsorbent for Extracting Hg (II) From Contaminated Water: Studies of Isotherm, Kinetics, and Mechanism. J Polym Environ 28(9):2498–2510
Wang J, Zhuang S (2017) Removal of various pollutants from water and wastewater by modified chitosan adsorbents. Crit Rev Environ Sci Technol 47(23):2331–2386
Aniagor CO, Afifi M, Hashem A (2021) Rapid and efficient uptake of aqueous lead pollutant using starch-based superabsorbent hydrogel. Polym Bull. https://doi.org/10.1007/s00289-021-03817-4
Hashem A, Aniagor C, Hussein D, Farag S (2021) Application of novel butane-1, 4-dioic acid-functionalized cellulosic biosorbent for aqueous cobalt ion sequestration. Cellulose 28:3599–3615
Hashem A, Aniagor CO, Afifi MAF, Abou-Okeil A, Samaha SH (2021) Synthesis of super-absorbent poly (AN)-g-starch composite hydrogel and its modelling for aqueous sorption of cadmium ions. Korean J Chem Eng. https://doi.org/10.1007/s11814-021-0856-7
Zhou Y, Fu S, Zhang L, Zhan H, Levit MV (2014) Use of carboxylated cellulose nanofibrils-filled magnetic chitosan hydrogel beads as adsorbents for Pb (II). Carbohydr Polym 101:75–82
Abdel-Halim ES (2013) Preparation of starch/poly(N,N-Diethylaminoethyl methacrylate) hydrogel and its use in dye removal from aqueous solutions. React Funct Polym 73(11):1531–1536
Kjeldahl JG (1883) Neue methode zur bestimmung des stickstoffs in organischen körpern. Zeitschrift für analytische Chemie 22(1)366–382
Hashem A, Afifi M, El-Alfy E, Hebeish A (2005) Synthesis, characterization and saponification of poly (AN)-starch composites and properties of their hydrogels. Am J Appl Sci 2(3):614–621
Asuquo E, Martin A, Nzerem P, Siperstein F, Fan X (2017) Adsorption of Cd (II) and Pb (II) ions from aqueous solutions using mesoporous activated carbon adsorbent: Equilibrium, kinetics and characterisation studies. J Environ Chem Eng 5(1):679–698
Aniagor C, Abdel-Halim E, Hashem A (2021) Evaluation of the aqueous Fe (II) ion sorption capacity of functionalized microcrystalline cellulose. J Environ Chem Eng 9(4):105703
Igwegbe CA, Oba SN, Aniagor CO, Adeniyi AG, Ighalo JO (2020) Adsorption of ciprofloxacin from water: a comprehensive review. J Ind Eng Chem 93:57–77
Kuang Y, Zhang X, Zhou S (2020) Adsorption of methylene blue in water onto activated carbon by surfactant modification. Water 12(2):587
Aniagor CO, Sokker H, Hashem A, El-Hamaki Y, El-Degwi M, Abdel-Halim E, Hashem A (2021) Equilibrium studies on the uptake of nitrate and phosphate ions using functionalized carbon cloth. J Radioanal Nucl Chem 329:1091–1102
Girish C, Murty VR (2016) Mass transfer studies on adsorption of phenol from wastewater using Lantana camara, forest waste. Int J Chem Eng. https://doi.org/10.1155/2016/5809505 (Article ID 5809505)
Pekař M (2020) Thermodynamic driving forces and chemical reaction fluxes; reflections on the steady state. Molecules 25(3):699
Prah A, Frančišković E, Mavri J, Stare J (2019) Electrostatics as the driving force behind the catalytic function of the monoamine oxidase a enzyme confirmed by quantum computations. ACS Catal 9(2):1231–1240
Wang J, Guo X (2020) Adsorption isotherm models: Classification, physical meaning, application and solving method. Chemosphere p. 127279
Langmuir I (1916) The constitution and fundamental properties of solids and liquids. Part I. Solids. J Am Chem Soc 38(11):2221–2295
Adamson AW, Gast AP (1967) Physical chemistry of surfaces, vol 150. Interscience publishers New York
Temkin M (1940) Kinetics of ammonia synthesis on promoted iron catalysts. Acta Physiochim URSS 12:327–356
Radushkevich M (1947) The equation of the characteristic curve of the activated charcoal USSR. Phys Chem Sec 55:331
Günay A, Arslankaya E, Tosun I (2007) Lead removal from aqueous solution by natural and pretreated clinoptilolite: adsorption equilibrium and kinetics. J Hazard Mater 146(1–2):362–371
Dąbrowski A (2001) Adsorption—from theory to practice. Adv Colloid Interface Sci 93(1–3):135–224
Dubinin M (1960) The potential theory of adsorption of gases and vapors for adsorbents with energetically nonuniform surfaces. Chem Rev 60(2):235–241
Wang G, Chang Q, Zhang M, Han X (2013) Effect of pH on the removal of Cr (III) and Cr (VI) from aqueous solution by modified polyethyleneimine. React Funct Polym 73(11):1439–1446
Jiang M, Yang W, Zhang Z, Yang Z, Wang Y (2015) Adsorption of three pharmaceuticals on two magnetic ion-exchange resins. J Environ Sci 31:226–234
Wang J, Guo X (2020) Adsorption kinetic models: Physical meanings, applications, and solving methods. J Hazard Mater 390:122156
Guo X, Wang J (2019) A general kinetic model for adsorption: theoretical analysis and modeling. J Mol Liq 288:111100
Aniagor CO, Afifi MA, Hashem A (2021) Modelling of basic blue-9 dye sorption onto hydrolyzed polyacrylonitrile grafted starch composite. Carbohydrate Polymer Technologies Applications 2:100141. https://doi.org/10.1016/j.carpta.2021.100141
Ho Y-S, McKay G (1999) Pseudo-second order model for sorption processes. Process Biochem 34(5):451–465
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aniagor, C.O., Afifi, M.A. & Hashem, A. Heavy metal adsorptive application of hydrolyzed corn starch. J Polym Res 28, 405 (2021). https://doi.org/10.1007/s10965-021-02772-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-021-02772-y