
Abstract
This work reports selective polyamidation of a diamine-phenol compound (3) in an ionic liquid (IL) and preparation of transparent composites from the obtained high performance polyamides (PA)s and modified γ-Al2O3 nanoparticles (mγNAl). The PAs were prepared selectively from polycondensation of compound 3 with several dicarboxylic acids in the mixture of (IL)/triphenylphophite(TPP). The fluorescent PAs (λem = 500 nm) exhibited glass transition (Tg) and 10 % weight loss temperatures (T10%) in the range of 188–325 °C and 447–511 °C, respectively. The structurally intercalated composites (NCPA)s were prepared from PAs and epoxide-functionalized γ-Al2O3 (mγNAl) via solution blending process. The strong chemical bonding between mγNAl and PA chains contributed to the enhanced thermal and mechanical properties of the composites. Also, the photoluminescence intensity increased with increasing mγNAl content. The PAs and composites were also tested for their extraction capability for environmentally deleterious metal ions from aqueous solutions either individually or in the mixture.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Ionic liquids (IL)s have received much attention in many areas of chemistry due to their useful properties such as solvent for polar and nonpolar species, catalyst and reusability [1, 2]. The ambient temperature ILs, mainly based on 1,3-dialkylimidazolium cations, have been extensively used in preparation of high performance polymers [3, 4]. Polyamides are excellent high performance polymers [5, 6], which have been modified structurally extensively to improve their processability [7–10]. The heteroaromatic rings, if they are chosen carefully, are likely to increase solubility of the polyamides without affecting thermal and mechanical properties to any great extent [11–13]. Also, introducing ether linkage and trifluoromethyl (CF3) group in polymer chain were found successful in altering crystallinity and intermolecular interactions and hence increase flexibility, solubility and transparency of polymer [14–16]. Application of inorganic nanoparticles (1–100 nm) as additives has improved general performance of polymers and has motivated increased researches and development in composites. The advantages in the mechanical properties of the nanocomposites, such as the higher modulus and yield stress, are usually accompanied by an increase in the melt viscosity and a change in the rheological behavior [17–19].
In this paper, selective synthesis of new PAs from a diamine-phenol compound in the mixture of IL/TPP is reported without using Py/NMP/LiCl which is required in the Yamazaki’s direct polycondensation. It is demonstrated that composite films can be readily achieved by heating a stable and homogeneous dispersion of the PA containing reactive phenolic hydroxyl and amine groups in the chains and epoxide-end capped γ-Al2O3 nanoparticles. The composites were characterized by using FT-IR, XRD, AFM and SEM techniques and their properties such as solubility, thermal, photophysical, mechanical and ability for removal of toxic heavy metal ions were investigated.
Experimental
Materials
All chemicals were purchased from Fluka and Merck Chemical Co. (Germany). Ammonium acetate, hydrazine monohydrate, 10 % palladium on activated carbon, 3-glycidyloxypropyl trimethoxysilane (GPTES), terephthalic acid, isophthalic acid, pyridine-2,6-dicarboxylic acid, 4,4′-sulfonyldibenzoic acid, adipic acid and sebacic acid were used as received. N-methyl-2-pyrrolidone (NMP), N,N-dimethylacetamide (DMAc), and pyridine (Py) were purified by distillation under reduced pressure over calcium hydride. γ-Al2O3 nanoparticles (γNAl) having an average size of 50 nm were purchased from Aldrich (Germany). 4,4′-dihydroxy benzil and 1,3-(propyl)2im]+ Br¯ were synthesized recently in our research laboratory [15].
Monomer synthesis
Synthesis of 1, 2-bis(4-(4-nitro-2-(trifluoromethyl)phenoxy)phenyl)ethane-1,2-dione (1). Into a 100 mL round-bottomed two-necked flask equipped with a magnetic stirrer bar and a reflux condenser, 4,4′-dihydroxy benzil (1.21 g, 0.005 mol) and 1-chloro-4-nitro-2-(trifluoromethyl)benzene (2.25 g, 0.01 mol) were dissolved in 10 mL DMAc, and potassium carbonate (1.38 g, 0.01 mol) was added to the solution. After 30 min of stirring at room temperature, the mixture was heated at 110 °C for 6 h and then was poured in 100 mL distilled water. The yellow precipitate was filtered off and dried in a vacuum oven at 80 °C. Yield: 93 % (2.9 g) and m. p: 157–160 °C. FT-IR (KBr) at cm−1: 3045 (aromatic C-H); 1621 (C = O); 1482 (C = C); 1534, 1353 (NO2); 1268 (C-O) stretching.1H NMR (400 MHz, DMSO-d 6,): δ7.41 (d, 4H, Ar-H, J = 8.0 Hz), 7.46 (d, 2H, Ar-H, J = 8.0 Hz), 8.08 (d, 4H, Ar-H, J = 8.0 Hz), 8.54 (dd, 2H, Ar-H, J = 8.0 Hz), 8.57 (d, 2H, Ar-H, J = 2.8 Hz). Elemental analysis calculated for C28H14F6N2O8: C, 54.19 %; H, 2.26 %; N, 4.52 % and found: C, 54.09 %; H, 2.37 %; N, 4.53 %.
Synthesis of 4- (4,5-bis(4-(4-nitro-2-(trifluoromethyl) phenoxy)phenyl)-1H-imidazole-2-yl) phenol (2). In a 500 mL round-bottomed two-necked flask equipped with a condenser, a magnetic stir bar and a nitrogen gas inlet tube, a mixture of 1.22 g (0.01 mol) 4-hydroxy benzaldehyde, 6.19 g (0.01 mol) compound 1, 5.39 g (0.07 mol) ammonium acetate and 50 mL glacial acetic acid was refluxed for 24 h. Upon cooling, the white precipitate was collected by filtration and washed with ethanol and water. The yield of the crude product was 6.6 g (91 %). The crude product was recrystallized from ethanol to afford white solid with melting point 150–154 °C. FT-IR (KBr disk) at cm−1: 3200–3452 (OH and NH imidazole), 3067 (C-H aromatic), 1611 (C = N), 1588 (C = C), 1532, 1350 (NO2), 1267 and 1230 (C-O-C).1HNMR (DMSO-d6, δ in ppm): 6.14 (d, 1H, Ar-H, J = 8.2 Hz), 6.38 (s, 1H, Ar-H), 6.71 (d, 1H, Ar-H, J = 8.2 Hz), 7.17–7.39 (m, 14H, Ar-H), 8.17–8.37 (m, 4H, Ar-H), 9.70 (s, 1H, OH phenol), 12.10 (s, 1H, NH imidazole ring). Elemental analysis calculated for C35H20F6N4O7: C, 58.17 %; H, 2.77 %; N, 7.76 % and found: C, 58.02 %; H, 2.85 %; N, 7.74 %.
Synthesis of 4- (4,5-bis(4-(4-amino-2-(trifluoromethyl)phenoxy)phenyl)-1H-imidazole-2-yl) phenol (3)nn To a 250 mL round-bottomed three-necked flask equipped with a dropping funnel, a reflux condenser and a magnetic stir bar, 6.62 g (0.01 mol) compound 2 and 0.2 g palladium on activated carbon (Pd/C, 10 %) were dispersed in 80 mL ethanol. The suspension solution was heated to reflux, and 8 mL of hydrazine monohydrate was added slowly to the mixture. After a further 5 h of reflux, the solution was filtered hot to remove Pd/C, and the filtrate was cooled to give white crystals. The product was collected by filtration and dried in vacuum oven at 80 °C. The yield of the reaction was 82 % (5.4 g), and the melting point was 144–147 °C. FT-IR (KBr disk) at cm−1: 3368–3472 (OH, NH2 and NH imidazole), 3047 (C-H aromatic), 1630 (C = N), 1597 (C = C), 1266 and 1212 (C-O-C). 1H NMR (DMSO-d6, δ in ppm): 5.45 (s, 2H, NH), 5.51 (s, 2H, NH), 6.80–6.85 (m, 6H, Ar-H), 6.89–6.97 (m, 6H, Ar-H), 7.43 (d, 2H, Ar-H, J = 7.6 Hz), 7.50 (d, 2H, Ar-H, J = 7.6 Hz), 7.86 (d, 2H, Ar-H, J = 7.6 Hz), 9.68 (s, 1H, OH phenol), 12.31 (s, 1H, NH imidazole ring). 13C NMR (100 MHz, DMSO-d6, δ in ppm): 111.09, 115.84, 116.92, (117.03, 117.15, 117.37 (C-C-CF3)), 119.06, (122.11, 122.42, 122.51, 122.63 (C-CF3)), 122.76 (CF3), 123.57, 123.97, 125.51 (CF3), 125.97, 126.76, 127.15, 128.12 (CF3), 128.65, 130.36, 131.17 (CF3), 136.13, 142.86, 146.11, 146.17, 146.52, (157.39, 157.54, 157.76 (C-C-CF3)), 158.12 and 158.43. Elemental analysis calculated for C35H24F6N4O3: C, 63.44 %; H, 3.63 %; N, 8.46 % and found: C, 63.37 %; H, 3.87 %; N, 8.42 %.
Polymer synthesis
Polyamides (PA)s were synthesized using two synthetic procedures:
-
(1)
By direct polycondensation of Yamazaki method [20] using mixture of TPP(1.20 mmol)/NMP(5 mL)/Py(1 mL)/LiCl(0.3 g), compound 3 (1 mmol, 0.54 g) and a dicarboxylic acid(1 mmol). An insoluble heterogeneous mixture was formed which was transformed into polymer film by pouring the mixture into a 9 cm-diameter glass culture dish and heating under vacuum at 80 °C for 1 h, 100 °C for 2 h, and 140 °C for 5 h to evaporate the solvent slowly. Polymer films were self-stripped off from the glass surface by soaking in water. The polymer films were further dried in a vacuum oven at 160 °C for 6 h.
-
(2)
By direct polycondensation using mixture of TPP (1.29 mmol), IL(0.70 g), compound 3 (1 mmol, 0.54 g) and a dicarboxylic acid (1 mmol) without using NMP/Py/LiCl. The homogeneous viscous solution (after 2.5 h at 110 °C) were precipitated in methanol, filtered and washed with hot water. The yields of PAs were in the range of 81–93 % after removal of low molecular weight fractions by washing with refluxing methanol for 24 h in a Soxhlet apparatus. The inherent viscosity (ηinh) of the resulting PAs, measured at a concentration of 0.5 g/dL in NMP at 25 °C, was between 0.52 and 0.91 dL g−1. The above procedure was used for the preparation of all PAs, Scheme 1.
Scheme 1 
Reaction scheme for selective polyamidation and synthesis PAs/mγNAl composites

-
PA1. This polymer was synthesized from compound 3 and terephthalic acid. Yield = 86 % and ηinh = 0.77 dL/g. FT-IR (KBr disk) at cm−1: 3200–3450 (OH, NH amide and NH imidazole), 3078 (C-H aromatic), 1674 (C = O amide), 1617 (C = N), 1555 (C = C), 1249 and 1211 (C-O). 1H NMR (DMSO-d 6, δ in ppm): 6.87–8.33 (m, 22H, Ar-H), 9.83 (s, 1H, OH phenol), 10.72 (s, 2H, NH amide), 12.89 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C43H26F6N4O5)n: C,65.15 %; H, 3.28 %; N, 7.07 %. Found: C, 65.02 %; H, 3.37 %; N, 7.09 %.
-
PA2. This polymer was synthesized from compound 3 and isophthalic acid. Yield 81 % and ηinh = 0.63 dL/g. FT-IR (KBr disk) at cm−1: 3260–3551 (OH, NH amide and NH imidazole), 3043 (C-H aromatic), 1658 (C = O amide), 1611 (C = N), 1518 (C = C), 1251 and 1217 (C-O).1H NMR (DMSO-d 6, δ in ppm): 6.67–8.11 (m, 22H, Ar-H), 9.82 (s, 1H, OH phenol), 10.71 (s, 2H, NH amide), 12.86 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C43H26F6N4O5)n: C,65.15 %; H, 3.28 %; N, 7.07 %. Found: C, 64.93 %; H, 3.42 %; N, 7.08 %.
-
PA3. This polymer was synthesized from compound 3 and 2,6-pyridinedicarboxylic acid. Yield 91 % and ηinh = 0.82 dL/g. FT-IR (KBr disk) at cm−1: 3280–3619 (OH, NH amide and NH imidazole), 3073 (C-H aromatic), 1668 (C = O amide), 1600 (C = N), 1515 (C = C), 1252 and 1217(C-O). 1H NMR (DMSO-d 6, δ in ppm): 6.93–8.47 (m, 21H, Ar-H), 9.93 (s, 1H, OH phenol), 10.76 (s, 2H, NH amide), 12.95 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C42H25F6N5O5)n: C,63.56 %; H, 3.15 %; N, 8.83 %. Found: C, 63.44 %; H, 3.29 %; N, 8.79 %.
-
PA4. This polymer was synthesized from compound 3 and 4,4′-sulfonyldibenzoic acid. Yield 87 % and ηinh = 0.60 dL/g. FT-IR (KBr disk) at cm−1: 3270–3580 (OH, NH amide and NH imidazole), 3074 (C-H aromatic), 1686 (C = O amide), 1612 (C = N), 1511 (C = C), 1253 and 1215 (C-O). 1H NMR (DMSO-d 6, δ in ppm): 7.14–8.57 (m, 26H, Ar-H), 9.98 (s, 1H, OH phenol), 10.81 (s, 2H, NH amide), 13.38 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C49H30F6N4O7S)n: C, 63.09 %; H, 3.22 %; N, 6.01 %. Found: C, 62.96 %; H, 3.40 %; N, 6.00 %.
-
PA5. This polymer was synthesized from compound 3 and adipic acid. Yield 92 % and ηinh = 0.47 dL/g. FT-IR (KBr disk) at cm−1: 3250–3530 (OH, NH amide and NH imidazole), 3049 (C-H aromatic), 1674 (C = O amide), 1616 (C = N), 1529 (C = C), 1248 and 1203 (C-O). 1H NMR (DMSO-d 6, δ in ppm): 1.67 (m, 4H, C-H), 2.33 (t, 4H, C-H), 6.72–8.09 (m, 18H, Ar-H), 9.78 (s, 1H, OH phenol), 10.77 (s, 2H, NH amide), 13.48 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C41H30F6N4O5)n: C,63.73 %; H, 3.89 %; N, 7.25 %. Found: C, 63.63 %; H, 4.06 %; N, 7.19 %.
-
PA6. This polymer was synthesized from compound 3 and sebacic acid. Yield 93 % and ηinh = 0.54 dL/g. FT-IR (KBr disk) at cm−1: 3260–3540 (OH, NH amide and NH imidazole), 3075 (C-H aromatic), 1661 (C = O amide), 1608 (C = N), 1513 (C = C), 1254 and 1202 (C-O). 1H NMR (DMSO-d 6, δ in ppm): 1.35 (m, 8H, C-H), 1.59 (m, 4H, C-H), 2.32 (t, 4H, C-H), 6.71–7.99 (m, 18H, Ar-H), 9.75 (s, 1H, OH phenol), 10.73 (s, 2H, NH amide), 13.40 (s, 1H, N-H, imidazole ring). Elemental analysis calculated for (C45H38F6N4O5)n: C,65.22 %; H, 4.59 %; N, 6.76 %. Found: C, 65.11 %; H, 4.83 %; N, 6.74 %.
Surface modification of γNAl particles (mγNAl)
Silane coupling agent 3-glycidoxypropyltriethoxysilane (GPTES), as shown in Scheme 1, was introduced to ensure good dispersion and improvement of interface interaction between γNAl particles and PAs. Surface modification of γNAl was carried out using a modified procedure [21]. 1 g γNAl in 30 mL ethanol/water (50/50, v/v) was sonicated in a 150 W ultrasonic water bath for 30 min. 1 g GPTES added to the above solution and the mixture was stirred at 80 °C for 4 h and then at room temperature for 24 h. The suspension was sonicated twice in methanol for 20 min to remove unreacted GPTES molecules, then mγNAl was separated by filtration and dried in a vacuum oven at 100 °C for 24 h. The mγNAl was kept under vacuum in order to protect the nanoparticles surface from moisture.
Preparation of mγNAl/PA composites (NCPA)s
0.1 g of a PA was dissolved in 20 mL dry NMP by stirring in an ultrasonic water bath. Then a certain amount of mγNAl particles (5, 10, 15 and 20 wt% based on weight of PA) was added to the resulting solution and the mixture was stirred for 30 min at 70 °C. After irradiation, 0.2 g K2CO3 was added and the mixture was continuously stirred at 80 °C for 5 h under nitrogen atmosphere. Finally, the mixture was cooled, filtered, and the obtained solid was washed with refluxing ethanol (soxhlet extraction) to remove any unreacted materials. The obtained composites were dried under reduced pressure at 60 °C for 12 h. Film preparation from the prepared NCPAs was carried out by stirring a certain amount of NCPA (5, 10, 15 and 20 wt%) in DMF in an ultrasonic water bath for 1 h to form a homogeneous dispersion. The mixture was further stirred for 24 h at 60 °C, forming a viscose solution which was casted into a flat-bottom glass plate. The casted composite was heated under nitrogen atmosphere at different temperatures of 70, 100, 200 °C for 1 h each and at 250 °C for 2 h, forming film of NCPAs with thicknesses of around 3 mm.
Measurements
1H NMR spectra were recorded on a 400 MHz BrukerAvance DRX instrument (Germany) using DMSO-d6 as solvent. FT-IR spectra were recorded using a Bruker Tensor 27 spectrometer on KBr pellets. Elemental analyses were performed using CHN-600 Leco elemental analyzer. The inherent viscosity (concentration 0.5 g/dL) was measured with an Ubbelohde suspended-level viscometer at 25 °C using NMP as solvent. Qualitative solubility was determined using 0.05 g of a polymer in 0.5 mL of solvent. The GPC measurements were conducted at 30 °C with a Perkin-Elmer instrument equipped with a differential refractometer detector, columns packed with a polystyrene/ divinylbenzene copolymer and DMF as fluent at a flow rate of 1 mL/min. Thermogravimetric analysis (TGA) and differential scanning calorimeter (DSC) was performed with the DuPont Instruments (TGA 951) analyzer and Perkin Elmer pyres 6 DSC, respectively, at a heating rate of 10 °C/min under N2, and for TGA also in air. Tg values were taken from the second heating scan after cooling from 400 °C. Ultraviolet–visible and fluorescence emission spectra were recorded on a Cecil 5503 (Cecil Instruments, Cambridge, UK) and Perkin-Elmer LS-3B spectrophotometers (Norwalk, CT, USA) (slit width = 2 nm), respectively, using a dilute polymer solution (0.20 g/dL) in DMSO. PAs films were prepared by dissolving 0.5 g PA in 5 mL DMF and the homogeneous solution was poured into a 9 cm-diameter glass culture dish, heated under vacuum at 50 °C for 2 h, 100 °C for 5 h, and 150 °C for 3 h to evaporate the solvent slowly. Polymer films were self-stripped off from the glass surface by soaking in water and then dried in a vacuum oven at 170 °C for 10 h. X-ray powder diffraction patterns were recorded by an X-ray diffractometer (GBC MMA instrument) with Be-filtered Cu K (1.5418 A°) operating at 35.4 kV and 28 mA with 2θ scanning range of 4–50° at a scan rate of 0.05° pers. FE-SEM was recorded on a Hitachi S4160 instrument. Branson S3200 (50 kHz, 150 W) ultrasonic bath was used for better dispersion of nanoparticles. Tensile properties of the polymers (strips of width = 5 mm, length = 30 mm and thickness = 30–50 μm) were obtained using MTS Criterion™ Universal Test Systems, with extension rate of 5 mm min−1and gauge length of 10 mm. Six samples were tested for each polymer and the average values are reported. For the moisture absorption measurement, 200 mg PA powder was dried at 120 °C for 8 h and then placed in an open space with a relative humidity of 80 %. The samples were weighed periodically over the course of 48 h. The solid–liquid extraction of Cr+3, Co+2, Zn+2, Pb+2, Cd+2, Hg+2, Mn+2, Ni2+, Cu2+, and Fe3+ as their nitrate salts was carried out at pH = 7–8. Approximately 50 mg of the polymer powder was shaken with 50 mL of an aqueous solution of the metal salt (initial concentration = 20 mg L−1) for a week at 25 °C. After filtration, the concentration of each metal cation in the liquid phase was determined by atomic absorption spectrophotometer (BRAIC WFX-130 AA). Direct information regarding the extraction percentage of metal ion by the adsorbent was obtained by using a calibration curve made from standard solutions of 5, 10, and 20 ppm for each metal salt. The amount of adsorbed ion was calculated using the following equation:
Where Q t is the amount of metal ion adsorbed on the adsorbent (mg g−1), C 0 and C A are the concentrations of metal ion in the initial solution and in the aqueous phase after adsorption, respectively (mg mL−1), V is the volume of the initial solution (mL) and w is the weight of the adsorbent (g). Selectivity coefficient of adsorption of metal ion was studied in the mixture by batch procedure. The distribution coefficient (k d ) of the metal ion was determined by the following equation:
The extraction selectivity, α, is the ratio of two distribution coefficients as shown in Eq. (3). The HgII cation was taken as the reference, as it has the highest distribution coefficient.
Result and discussion
Synthesis and characterization of compound (3)
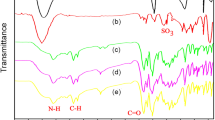
The FT-IR spectrum of compound 2 in Fig. 1 shows two characteristics absorption bands at 1530 and 1350 cm−1 due to asymmetric and symmetric –NO2 stretching vibrations. After reduction, new absorption bands were observed at 3470 and 3360 cm−1 due to N–H stretching. The 1H NMR spectrum of compound 3 in Fig. 2a shows the characteristic signals of two different kinds of protons: NH2 protons at 5.45 and 5.51 ppm and O–H and N-H of hydroxyl groups and imidazole ring at 9.68 and 12.31 ppm, respectively. The 13C NMR spectrum of compound 3 showed 24 different carbon atoms. There are quartet peaks in 13C NMR spectrum because of the heteronuclear 13C-19 F coupling. The large quartet centered at about 123 ppm is due to the –CF3 carbons. The CF3- attached carbon also shows a clear quartet centered at about 122 ppm with a smaller coupling constant of about 32 Hz due to two-bond C–F coupling. These quartet signals corroborate the presence of CF3 group in the synthesized compounds. Thus, all the spectroscopic data obtained by 1H and 13C NMR techniques were in good agreement with the expected chemical structures.

FT-IR spectra of γ-Al2O3, GPTES- γ-Al2O3, compound 3, PA3 and NCPA3-10 %

1H NMR spectrum of compound 3 (a) andPA1 (b) in DMSO-d6
Synthesis and characterization of PAs and composites
The ambient temperature ILs, especially those based on 1,3-dialkylimidazolium cations, have gained considerable interest as promising alternative green solvents in polymer synthesis [4, 21]. In this paper, selective polyamidation is reported for an aromatic diamine-phenol compound 3 with several commercial dicarboxylic acids in imiazolium-based IL, [1,3-(propyl)2im]+ Br¯. The polycondensation reaction proceeded selectively and efficiently in the mixture of IL/TPP without using NMP/Py/LiCl mixture which is required when using direct polycondensation of Yamazaki method [20]. The highest yield and viscosity were obtained in [1,3-(Pr)2im]+ Br¯ after 2.5 h at 110 °C. The weight average molecular weights (Mw) of PAs measured by GPC were in the range of 42,300–58,500 g/mol with the molar mass dispersity in the range of 1.49–1.87, Table 1. The relatively high molecular weight can be due to the use of IL as solvent at high temperature with low vapor pressure, which facilitates the elimination of the byproduct and thus shifting the equilibrium. When polymerization was preceded in TPP/NMP/Py/LiCl mixture, a dark color and insoluble material was obtained indicating formation of a thermoset material through multifunctional reactions between carboxylic acid, amine and phenolic hydroxyl groups. This demonstrates the beneficial effect of IL in the synthesis of thermoplastic PAs selectively from a diamine-phenol, in addition to other useful factors such as the non-volatility of ILs, low reaction time, no environmental pollution, and no need to remove chemicals such as NMP, LiCl and Py which are essential for conventional direct polycondensation. The 1H NMR spectrum of the PA1, Fig. 2b, shows the amide N-H proton at 10.72 ppm, imidazole N-H proton at 12.89 ppm and phenolic O-H proton at 9.83 ppm. The signals of aromatic and aliphatic protons appeared in the range of 6.67–8.57 ppm and 1.35–2.33 ppm, respectively.
PA/mγNAl composites were successfully prepared through formation of chemical bond between organic and inorganic phases. First, the γNAl was grafted with GPTES (mγNAl) through condensation of hydroxyl groups (−OH) on the surface of activated γNAl with the hydrolysable methoxy groups (−OCH3) of GPTES. FT-IR spectrum of γNAl in Fig. 1 shows the absorption bands at 545 and 788 cm−1 which are related to the aluminium ions in octahedral and tetrahedral environments. FT-IR spectrum of mγNAl in Fig. 1 presents the characteristic absorption bands at: 1100 cm−1related to the vibration of Si-O link, 3400 cm−1 to the stretching vibration of -OH on the surface of γNAl, 1380 cm−1 to the bending and at 2864–2917 cm−1 to the stretching vibrations of C-H of organic methylene. These results are in agreement with the results reported by other researcher [22]. In addition, the characteristic absorption band at 1097–1198 cm−1 broadens which reveals formation of a new absorption band of Al-O-Si due to the interaction between siloxane and γNAl. The above interpretations are enough to manifest that GPTES is bound on the surface of γNAl. As shown in Scheme 1, the formation of chemical bond between the PA and mγNAl is suggested as a result of the reactions between the epoxide groups of mγNAl and the hydroxyl and amine functional groups in the PA chains. FT-IR spectrum of the PA/mγNAl composite, Fig. 1, exhibits the characteristic absorption bands of the amide group in the regions of 3386 cm−1 (N-H stretching) and 1668 cm−1 (C = O stretching), and Al-O-Si band at 1097–1198 cm−1. Scattering is a powerful tool to access the bulk structure in a nondestructive way. X-ray diffraction (XRD) has been used to investigate the nature of PA/mγNAl composite with respect to neat PAs. XRD patterns for the neat PA3, γNAl, mγNAl, and PA/mγNAl composites (NCPA1-5 %) are shown in Fig. 3a. According to these patterns, the PA3 is totally amorphous in nature which does not show any sharp diffraction peaks, while XRD pattern of the PA/mγNAl composite expressing that the crystal structure of the γNAl was not altered and the peak intensity depends on the amount of GPTES and PA.

a XRD patterns of γ-Al2O3, GPTES- γ-Al2O3, PA3 and NCPA3-10 %, b UV–vis and fluorescence spectra of PAs, composites and composite/Hg+2 complex, c DSC curves of PAs, composites and mixture of mγNAl with PA1 and d TGA curves of PAs, γNAl, mγNAl and NCPA1-10 %
Properties of PAs and composites
Photophysical properties
The UV–vis absorption and fluorescence emission spectra of PAs and composites films are shown in Fig. 3b and the extracted data are listed in Table 1. The absorption spectra of PAs in films and in dilute NMP solutions (0.2 g/dL) were nearly identical with λab = 311–318 nm which are related to the π → π* transition of aromatic rings. The photoluminescence (PL) quantum yields (Φf) of dilute PAs solutions (0.2 g/dL) were determined in NMP using excitation wavelength of 320 nm. A 0.10 N solution of quinine in H2SO4 (Φf = 0.53) was used as described in the literature [23]. The PAs solutions and thin films showed broad fluorescent emission spectra with λem = 451–504 nm and quantum yields of Φ f = 7–30 % (Table 1). The aliphatic PA5 and PA6 exhibited blue emission (λem = 451–466 nm) with the highest Φf values of 25 and 30 %, respectively. The aromatic PAs exhibited green emission (λem = 491–504 nm) and Φf in the range of 7–11 %. The blue shift and higher PL intensity of aliphatic PAs can be due to the effectively reduced conjugation and capability of charge transfer complex formation by aliphatic diacids.23. The composites films showed absorption at λab = 312–323 nm, emission at λem = 452–508 nm and Φf in the range of 11–36 % (Fig. 3b and Table 1). The composites prepared from the aliphatic PAs (NCPA5-5 % and NCPA6-5 %) showed blue shift with Φf of 32 and 36 %, respectively, in comparison with those prepared from the aromatic PAs which showed green shift with Φf in the range of 11–17 %.
Thermal properties
TGA and DSC methods were applied to evaluate thermal stability of the PAs and mγNAl/PA composites. Figure 3c shows DSC curves of the PAs, neither crystallization exotherm nor melting endotherm were observed in the range of 35–400 °C, so that these PAs were considered to be essentially amorphous. The amorphous nature of the PAs can be attributed to their bulky pendant group which decreased the inter-chain interaction resulting in loose polymer chain packaging and aggregates. The increased rotational barrier caused by the bulky pendant and inter-chain interactions enhanced Tg values of the PAs. The Tg values of the PAs (181–317 °C, Table 2) also depend on the stiffness of diacid component in the polymer chain. The PA5 and PA6 obtained from aliphatic dicarboxylic acids showed the lowest Tg values (196 °C and 181 °C, respectively) compared to the other PAs derived from the aromatic diacids. Also, Tg values of the composites showed dependence on the amount of mγNAl particles. As shown in Table 2, Tg increased about 20 °C when the composite contains 5 wt% mγNAl, and Tg was not observed up to 400 °C when composite was prepared with 10 wt% mγNAl or more. This can also indicate the formation of cross-links as a result of reaction between phenolic hydroxyl group in PA chain and epoxide group in mγNAl. Previous studies on the adduct formation between 1-substituted imidazole [24, 25] and triazole [26] with the epoxide group suggested that reaction between more basic pyridine-type nitrogen with the epoxide group leads to a zwitter ionic structure, as shown in Scheme 1. DSC technique was also used to show the occurrence of chemical reaction between epoxide groups of mγNAl and functional groups in PA chains when composite prepared by solution blending at 70 °C. Therefore, mixture of PA1 and mγNAl was heated in the DSC sample pan and, as can be seen in Fig. 3c, a sharp exothermic peak appeared in the temperature range of 80–100 °C. While composites prepared by the solution blending at 70 °C, also shown in Fig. 3c, did not show exothermic peak in the region of 80–100 °C. The absence of exothermic peaks in the DSC curves of the composites indicates that epoxide ring in mγNAl particles reacted with the amine of imidazole ring and phenolic hydroxyl of PA chains during solution blending at 70 °C. The above interpretations, and also the insolubility of composites in organic solvents, are enough evidences to suggest that mγNAl is chemically linked with the PA chains. Figure 3d presents TGA curves of the PAs and the corresponding T10% in N2 (436–521 °C) and air (396–475 °C) are listed in Table 2. Char yield (CR) was used as a criteria for evaluating the limiting oxygen index (LOI) of the polymers using equation LOI = 17.5 + 0.4CR. In general, when the LOI of a polymer is higher than 26 % it is considered not to be flammable. For these PAs, LOI values were calculated based on their char yields at 800 °C. Due to rigid structure of the aromatic diacids, thermal stability of the aromatic PAs in terms of T10% values is higher than thermal stability of the aliphatic PAs (Table 2). Figure 3d also shows TGA curves of the γNAl, mγNAl and NCPA1-10 %. T10% value of PA1/10 wt% mγNAl composite (NCPA1-10 %) increased from 507 to 524 °C and its char yield increased from 58 to 65 % (Table 2). It is clear that the presence of mγNAl and its chemical interactions with the polymer chains shifted the decomposition temperature towards higher temperatures indicating enhanced thermal stability of the composite.
Solubility and moisture absorption
The solubility behavior of these PAs and mγNAl/PA composites was qualitatively tested in various organic solvents. The PAs prepared in the mixture of TPP/IL exhibited excellent solubility in polar aprotic solvents such as NMP, DMAc, DMF, DMSO and even in less polar solvents like pyridine, THF, and m-cresol, while that (PA1′″) prepared in the mixture of TPP/NMP/Py/LiCl did not dissolve in these solvents. In the previous reports, comparable aramids with almost the same backbone structure showed very less solubility in organic solvents [27], or were practically insoluble [28]. The good solubility of these PAs should be due to the presence of bulky heterocyclic pendant, their amorphous nature and unsymmetrical structure where solvent molecules can penetrate inside the chains and interact with the polar groups of the polymer chains such as phenolic hydroxyl and -CF3 groups, and ether and amide linkages. However, the PA5 and PA6 showed solubility in THF and m-cresol due to the presence of the aliphatic units instead of the rigid phenyls. Composite of the aliphatic PA6 with 5 wt% mγNAl was insoluble in the highly polar solvents at room temperature but showed little solubility when heated at 60 °C. This showed that 5 wt% mγNAl was not enough to form a complete cross-linked network with the polymer chains. However, composites containing more than 5 wt% mγNAl did not dissolve in these highly polar solvents and this can also be an evidence for formation of chemical bond between the PA chains and epoxide groups of mγNAl. The absorbed water diminishes the Tg and influences the mechanical and electrical properties, but in the membrane technology, greater water uptake implied better performance. Regarding these PAs which are partially hydrophilic and with bulky pendant which increases the free volume, their polar amide and free hydroxyl groups interact effectively with water molecules. As can be seen, the relative moisture intake by these PAs ranged from 8.35to 11.21 %.
Mechanical properties
The mechanical properties of these PAs and representative mγNAl/PA composite were measured in the tensile mode and the results are summarized in Table 2. The PAs showed tensile strength in the range of 51-83 MPa with elongation to break of 12.7–24.0 % and the Young’s modulus of 1.49–2.21GPa. While PA1″′, prepared by method 1 in the synthesis section, was a brittle polymer with very low elongation (1.19 %) and high Young’s modulus (5.47GPa) (Table 2). The PA3 showed the highest tensile strength and toughness due to the higher molecular weight in comparison with the other PAs. The PA5 and PA6 showed the lowest strength due to the presence of the aliphatic units in their backbones and also due to their lower molecular weights in comparison with the other PAs. As can be seen in Table 2, the noticeable trend of the tensile strength of specimens increased when increasing the mγNAl content. For example, the tensile strength of PA1 increased from 83 MPa to 119PMa for NCPA1-20 %, and simultaneously, the tensile elongation decreased from 23 to 9 %. As mentioned before, the enhancement of tensile strength corresponds to the reinforcing role of γNAl in the composite. It might also be ascribed to the presence of interfacial adhesion between mγNAl and PA phases. As it is known, elongation at break is a crucial parameter to judge the plasticity or ductility of the polymer. Generally, the addition of inorganic particles into the polymer matrix, and also in this case due to the formation of chemical interactions between the two phases, the elongation at break decreases.
Extraction of metal cations from aqueous solution
As these PAs are insoluble in water and showed high moisture absorption, thus only PA3 and mγNAl20 %/PA3 composite were used in solid–liquid extraction. Extraction of the metal ions such as HgII, CdII, NiII, CoII, CuII, CrIII and FeIII as their nitrate salts was carried out in the aqueous solutions at pH 7, either individually or in the mixture. The results for distribution coefficient and selectivity in the mixture of metal ions in solution are shown in Table 3. As can be seen in this table, the adsorption of the tested cations on the mγNAl20 %/PA3 composite increased in comparison with the adsorption level by the PA3. Also, the selective adsorption of HgII cation was the highest either in the mixture or individually. Therefore, it is clear that the quantitative separation of HgIIcation from the rest of the cations is possible. The extraction level of these cations especially HgII from the aqueous solution by these PAs and composites, points to the future applications in the fields of membrane preparation for cation transport, water decontamination and in sensing materials, among other applications.
Morphology and dispersibility of GPTES-γAl2O3
In order to examine the surface morphology and nanoparticles distribution within the composites, SEM and AFM analysis were conducted. Typical surface SEM images of the same magnification of neat γAl2O3 particles, GPTES-γAl2O3 (mγAl2O3), and mγAl2O3/PA3 composite are illustrated in Fig. 4a–c, respectively. It can be observed that the size and apparent morphology of the γAl2O3 particles has changed drastically as its surface has been covered chemically with organic phases. Figure 4c also indicates that epoxide-functionalized mγAl2O3 particles have strongly adhered interfacial to the functional moieties of the PA matrix. Figure 4d and e shows AFM images of mγAl2O3 and mγAl2O3/PA3 composite. It can be seen that the surface roughness is rather small for the case of mγAl2O3with an average size of 92 nm, while the NCPA3-10 % sample shows rougher surface of many hills and valleys. Also these images show that there is a good dispersion of mγAl2O3 nanoparticles in the polymer matrix, where sharp points are referred to Al2O3 nanoparticles.

FESEM images of (a) γ-Al2O3, (b) GPTES- γ-Al2O3, (c) composite (NCPA3-10 %), AFM images of GPTES- γ-Al2O3 (d) and NCPA3-10 % (e)
Conclusion
In this study, photo luminescent and high-performance organosoluble PAs have been synthesized selectively from an aromatic diamine-phenol compound in IL/TPP media without using NMP/Py/LiCl mixture. This procedure showed the advantages such as selectivity, high yields, shorter reaction time, and without using volatile chemicals and no need for the removal of chemicals such as NMP, LiCl and Py. The prepared PAs with relatively high Mw showed excellent solubility in organic polar solvents and high thermal stability in terms of Tg (181–317 °C) and T10% values (396–452 °C in air). Therefore, such modifications can be used as an effective strategy for improving properties and enhancing processability of the aramids. These functionalized PAs were used in the preparation of reinforced composites with the epoxide-end capped γ-Al2O3 nanoparticles. As compared with the pure PAs, the mγNAl/PA composites are significantly strengthened as a result of interfacial reaction of epoxide groups of mγNAl with functional groups in PA chains. The results also demonstrated that these PAs and particularly their composites can act efficiently as chelating agent for heavy metal ions.
References
Wasserscheid P, Welton T. Ionic Liquids in Synthesis. Edited by Copyright © 2002 Wiley- VCH Verlag GmbH & Co. KGaA
Lu J, Yan F, Texter (2009) J Prog Polym Sci 34:431–448
Lozinskaya EI, Shaplov AS, Vygodskii YS (2004) Eur Polym J 40:2065–2075
Mallakpour S, Taghavi M (2008) Polymer 49:3239–3249
García JM, García FC, Serna F, de la Peña JL (2010) Prog Polym Sci 35:623–686
Gaudiana RA, Minns RA, Sinta R, Weeks N, Rogers HG (1989) Prog Polym Sci 14:47–89
Ghaemy M, Aghakhani B, Taghavi M, AminiNasab SM, Mohseni M (2013) React Funct Polym 73:555–563
Taghavi M, Ghaemy M, AminiNasab SM, Hassanzadeh M (2013) Polymer 54:3828–3840
Liou GS, Hsiao SH, Ishida M, Kakimoto M, Imai Y (2002) J Polym Sci A Polym Chem 40:2810–2818
Liu YL, Li SH, Lee HC, Hsu KY (2006) React Funct Polym 66:924–930
Ghaemy M, AminiNasab SM (2010) React Funct Polym 70:306–313
Ghaemy M, Alizadeh R (2011) React Funct Polym 71:425–432
Ghaemy M, AminiNasab SM, Taghavi M, Hassanzadeh M (2012) J Macromol Sci Pure Appl Chem 49:772–783
Ghaemy M, Hasheminasr F, Alizadeh R, Taghavi M (2012) Macromol Res 20:614–622
Ghaemy M, Hassanzadeh M, Taghavi M, AminiNasab SM (2012) J Fluor Chem 142:29–40
Ghaemy M, Sharifi S, AminiNasab SM, Taghavi M (2013) Polym Bull 70:1125–1142
Cerneaux S, Zakeeruddin SM, Pringle JM, Cheng YB, Grätzel M, Spiccia L (2007) Adv Funct Mater 17:3200–3206
Bazzar M, Ghaemy M, Alizadeh R (2012) Polym Degrad Stab 97:1690–1703
Bazzar M, Ghaemy M (2013) Compos Sci Technol 86:101–108
Yamazaki N, Matsumoto M, Higashi F (1975) J Polym Sci A Polym Chem 13:1373–1380
Seckin T, Vural S, Koytepe S (2010) Polym Bull 64:115–126
Jian Z, Yu J, Guo ZX (2005) Macromol Chem Phys 206:1558–1567
Liou GS, Chang CW (2008) Macromolecules 41:1667–1674
Ricciardi F, Romanchick WA, Jouille MM (1983) J Polym Sci Polym Chem Ed 21:1475
Ghaemy M, Sadjady S (2006) J Appl Polym Sci 100:2634
Ryu BY, Emrick T (2011) Macromolecules 44:5693
Ge Z, Yang S, Tao Z, Liu J, Fan L (2004) Polymer 45:3627–3635
Feng K, Hsu FL, Van DerVeer D, Bota K, Bu XR (2004) J Photochem Photobio A 165:223–228
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Taghavi, M., Ghaemy, M., Nasab, S.M.A. et al. Polyamidation of a diamine-phenol compound in ionic liquid: preparation and properties of polyamides/epoxide-functionalized γ-Al2O3 composites. J Polym Res 22, 12 (2015). https://doi.org/10.1007/s10965-014-0645-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-014-0645-7