
Abstract
Two novel π-conjugated monomers, 8,11-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2-b] quinoxaline (OTAQ) and 8,12-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2,5]thiadiazolo[3,4,i]quinoxaline (OTATQ), were synthesized. These polymers contains on type electron donating unit 3-octylthiophene and two type of electron withdrawing unit that acenaphthyl-quinoxaline in OTAQ and acenaphthyl-thiadiazolo-quinoxaline in OTATQ, respectively. Electrochemical polymerization of the monomers were carried out in acetonitrile/dichloromethane soluvent mixture containing tetra-n-butylammonium hexafluorophosphate and electrochromic properties of polymers are described in this paper. Furthermore, the effects of structural difference on electrochemical redox behavior and spectroelectrochemical properties of the two resulting polymers were examined. The results showed that an anodic wave at +0.56 V vs Ag wire pseudo-reference electrode corresponding to the monomer OTAQ oxidation was observed, and an anodic wave at +0.91 V was observed in oxidation of OTATQ due to it contains stronger electron withdrawing unit in which contain thiadiazolo structure. The UV–vis-NIR spectra analysis revealed that the POTAQ film has two absorbance bands centered at 694 nm and longer than 1,600 nm and the POTATQ film has an absorbance at 1,014 nm, respectively. The band gaps of these polymers, defined as the onset of the absorption band at 694 nm for POTATQ and the absorption band at 1,014 nm for POTATQ, were determined as 1.06 eV for POTAQ and as 0.76 eV for POTATQ. The electrochromic results showed that the two polymer films revealed about 30 % optical contrast in the NIR region with low response time (5 s for POTAQ and 10 s for POTATQ).
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
By definition an electrochromic material have the possibility of changing its color reversibly when it is placed in an applied electric field. Electrochromic materials can be classified in two main types depending on their chemical structure. The first type is an inorganic material which has at least one colored and one bleached state such as WO3 [1], NiO2 [2] and TiO2 [3]. These materials are used in smart windows and optical shutter. The second type are organic materials that including organic dyes and conjugated polymers. The organic dyes are organic small molecules such as bipyridiliums which are a class of materials that are transparent in the stable dicationic state and would be highly colored upon one-electron reduction. Conjugated polymers have attracted wide attention due to their ease of process ability, rapid response times, and high optical contrasts and the ability to modify their structure to create multicolor electrochromic device [4–8]. Conducting polymer generation follows via a mechanism that is believed to involve either radical-cation/radical-cation coupling or attack of radical-cation on neutral monomer. In their oxidized states, conducting polymers are ‘doped’ with the counter anions (p-doping) and shows a delocalised p-electron band structure. The energy gap between the highest occupied p electron band (valence band) and the lowest unoccupied band (the conduction band) determining the intrinsic optical properties of these materials [9]. So, conjugated polymers with low-bandgap obtained by electrochemical polymerization of suitable redox active monomers are future materials for electrochromic devices because of their potential ability in visible, IR and microwave electrochromics. This type of polymers can be used in photoelectric devices such as electrochromic displays [10], vision systems or smart windows [11].
In recent years, many investigations on the design and synthesis of low-band gap electrochromic polymers have been the major attractive area and a most successful and flexible strategy to design small band gap polymers involves the alternation of electron-rich and electron deficient units along the polymer chain [12–15]. Some of this type polymers, quinoxaline units were used as acceptor unit and some works reported that low-bandgap polymers containing quinoxaline units as acceptor unit performed good electrochromic ability [16–27]. These polymers generally show a variety of excellent properties such as distinct optical changes throughout the visible region, fast switching time, low-voltage electrochromic (the voltage lower than 1.3 V).
In designing low-band gap conjugated polymers, changing the nature and the position of the heteroatom on the donor or acceptor units is an effective strategy due to the heteroatom plays an important role in controlling the electron withdrawing capabilities of electron acceptor unit [19]. So, in this study we synthesized two monomers, 8,11-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2-b]quinoxaline and 8,12-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2,5]thiadiazolo[3,4,i]quinoxaline, to obtain two novel polymers with low-band gap (Chart 1). These two polymers synthesized by electrochemical polymerization and the spectroelectrochemical and switching properties of the resulting polymers were investigated. We also discussed the influence of thiadiazolo ring to electrochemical and electrochromic properties of these polymers.

Chemical structure of the synthesized polymers
Experimental
Chemical
All chemicals were purchased from Shanghai Boka Chemical co. and used as received unless otherwise noted. Drytetrahydrofuran (THF) was distilled over sodium with addition of benzophenone. 4,7-dibromo-5,6-dinitro-benzo[1,2,5]thiadiazole (M2) was synthesized according to literature procedurs [28]. The tetra-n- butylammonium hexafluorophosphate (TBAPF6) electrolyte dissolved in the solvent mixture of dichloromethane (DCM) and acetonitrile (ACN) (95/5, v/v) and was used as supporting electrolyte solution in electrochemical polymerization and spectroelectrochemistry analysis process.
Apparatus
A Zahner-Zennuim electrochemical workstation was used for all electrochemical studies. Electrochemical polymerization was performed in a three-electrode cell consisting of an indium tin oxide doped glass slide (ITO) as the working electrode, platinum wire as the counter electrode, and Ag wire as the pseudo-reference electrode. Spectroelectrochemistry spectra and electrochromic switching were taken with a Shimadzu UV-3600 UV–vis-NIR Spectrophotometer linked with a MCP-1 Potentiostat. 1HNMR spectra were recorded in CDCl3 on VARIAN INOVA-400 spectrometer (400 MHz). Chemical shifts were given in ppm downfield from tetramethylsilane.
Experimental procedures
The synthetic routes toward the monomers are shown in Fig. 1.

Schematic route for the synthesis of monomers
Preparation of tributyl(4-octylthiophen-2-yl)stannane (M1)
Synthesis of tributyl(4-octylthiophen-2-yl)stannane was carried out by following well-established literature procedures [29, 30] with slight modifications. 3-octylthiophene (39.00 g, 200.0 mmol) dissolved in anhydrous THF (150 ml) and butyl-lithium in hexane (2.5 mol/L, 80 ml, 200.0 mmol) was slowly added to the solution at −80 °C. After which the mixture was stirred under nitrogen for 1.5 h, the temperature of the mixture was natural increasing to room temperature in 2 h and stirred for another 3 h. After that the mixture was cooled to −80 °C and tributylchlorostannane (60 ml, 220.0 mmol) was added dropwise. The mixture was stirred at −80 °C for additional 3 h, followed by quenching with 2 M aqueous sodium hydrogen carbonate (200 ml). After that the organic solvent was removed under reduced pressure and the remained solution was extracted with diethyl ether. The diethyl ether solution was washed several times with aqueous sodium carbonate and brine, and dried over MgSO4. After removing the solvents, the residue was purified on a column of neutral alumina (hexane as eluent), evaporated and further purified by Kugelrohr distillation to give the desired product as yellow oil (95.3 g, 98 %). 1H NMR (400 MHz, CDCl3) δ: 7.18 (d, J = 2.8 Hz, 1H); 6.96 (d, J = 1.2 Hz, 1H);2.66 ~ 2.62 (t, 2H); 1.61 ~ 1.52 (m, 8H); 1.42 ~ 1.28 (m, 16 h); 1.11 ~ 1.06 (m, 6H); 0.92 ~ 0.86 (m, 12H).
Preparation of 4,7-bis(4-octylthiophene-2-yl) - [1,2,5] benzothiadiazole (M3)
To the solution of 4,7-debromo- [1,2,5] benzothiadiazole (2.91 g, 10.0 mmol) in anhydrous toluene (100 ml) was added M1 (10.60 g, 22.0 mmol) and was stirred at room temperature under nitrogen for 30 min. Then Pd(PPh3)Cl2 (0.14 g, 0.2 mmol) was added to the solution and the mixture was refluxed for 6 h. Then the solvent was removed under reduced pressure and 10 % HCl solution was added after the remaining browed oil was cooled to the room temperature. After stirred for 1 h, the solution was extracted with CH2Cl2, followed by washing with aqueous sodium carbonate and brine. The organic solvent was removed under reduced pressure after the solution was dried over MgSO4, and was purified on a column silica gel using CHCl3: petroleum ether (60 ~ 90) = 1:1 as the eluent to give the desired product as orange crystal (4.76 g, 91 %). 1H NMR (400 MHz, CDCl3) δ: 7.97 (d, J = 1.2 Hz, 2H); 7.82 (s, 2H); 7.03 (d, J = 3.2 Hz, 2H); 2.71 ~ 2.67 (t, 4H); 1.72 ~ 1.68 (m, 4H); 1.39 ~ 1.25 (m, 20H); 0.90 ~ 0.86 (m, 6H).
Preparation of 4,7-bis(4-octylthiophene-2-yl)-5,6-dinitro-[1,2,5]benzothiadiazole (M4)
4,7-bis(4-octylthiophene-2-yl)-5,6-dinitro-[1,2,5]benzothiadiazole (M4) was synthesized using M2 as raw material by the same procedure used for the synthesis of M3. The crude product was purified on a silica gel column using CHCl3: petroleum ether (60 ~ 90) = 1:1 as the eluent to give the desired product as red-orange crystal (94 %). 1H NMR (400 MHz, CDCl3) δ: 7.32 (d, J = 2.8 Hz, 4H); 2.67 (t, 4H); 1.66 ~ 1.61 (m, 4H); 1.35 ~ 1.28 (m, 20H); 0.90 ~ 0.86 (m, 6H).
Preparation of 8,11-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2-b]quinoxaline (OTAQ)
To a solution of M3 (524 mg, 1.0 mmol) in acetic acid (50 ml), zinc dust (1.31 g, 20.0 mmol) and a few drops of water was added the solution, the reaction mixture was kept at 60 °C for 1 h and the color turned white-pink. The solution was filtered twice and acenaphthylenequinone (200 mg, 1.1 mmol) was added to the pink solution, then the solution was stirred for 72 h at room temperature, after which it was poured into water and was extracted by CHCl3 and the organic phase was dried over MgSO4. After removing the organic solvents, the crude product was purified on a silica gel column (CHCl3: petroleum ether(60 ~ 90 °C) = 1:2 as eluent) to give the desired product OTAQ as orange powder (540 mg, 84 %). 1H NMR (400 MHz, CDCl3) δ: 8.47 (d, J = 6.8 Hz, 2H); 8.13 (d, J = 2.0 Hz, 2H); 8.10 (d, J = 2.4 Hz, 2H); 7.87 (t, 2H); 7.78 (s, 2H); 7.16 (s, 2H); 2.73 (t, 4H); 1.77 ~ 1.71 (m, 4H); 1.57 ~ 1.25 (m, 20H); 0.91 ~ 0.87 (m, 6H).
Preparation of 8,12-bis(4-octyl-2-thienyl)-acenaphthyl-[1,2,5]thiadiazolo[3,4,i]quinoxaline (OTATQ)
A mixture of M4 (614 mg, 1.0 mmol) and fine iron powder (560 mg, 10.0 mmol) in acetic acid (50 ml), was stirred for 4 h at 100 °C. The reaction mixture poured into water after cooling to room temperature and extracted with diethyl ether, dried with MgSO4. After the solvent was removed under reduced pressure, acetic acid (50 ml) and acenaphthylenequinone (200 mg, 1.1 mmol) were added to the residue and stirred at room temperature for 72 h. The reaction mixture poured into water and extracted with CHCl3, and the organic phase washed aqueous sodium carbonate and brine. The organic solvent removed after the solution dried over MgSO4, and was purified on a silica gel column (CHCl3: petroleum ether (60 ~ 90) = 1:1 as eluent) to give the desired product as blue solid (88 %). δ: 8.83 (d, J = 1.2 Hz, 2H); 8.28 (d, J = 6.8 Hz, 2H); 8.02 (d, J = 8.4 Hz, 2H); 7.74 (t, 2H); 7.26 (d, J = 10.0 Hz, 2H); 2.76 (t, 4H); 1.80 ~ 1.74 (m, 4H); 1.49 ~ 1.25 (m, 20H); 0.92 ~ 0.88 (m, 6H).
Results and discussion
Electrochemistry
We attempted to carry out the electrochemical studies in several different supporting electrolyte such as NaClO4/LiClO4, tetrabutylammonium tetrafluoroborate (TBABF4), tetrabutylammonium perchlorate (TBAC) and tetra-n-butylammonium hexafluorophosphate (TBAPF6). The LiCO4/NaCIO4 mixture electrolyte cannot was used in electrochemical experiment due to the electrolyte are soluble in ACN or DCM/ACN solvent mixture when the DCM volume fraction less than 30 %, but the monomers are insoluble in such solvent mixture. Although the monomer and the LiCO4/NaCIO4 mixture are soluble in THF, but the electrochemical behavior of monomers were poor and cannot obtain good films. The electroactivity of monomers were examined in DCM solution containing 0.1 M TBABF4, TBAC and TBAPF6, respectively. The results showed that the electrochemical behaviors of monomers were poor when used TBABF4 and TBAC, but well in DCM solution containing TBAPF6. So we mainly investigated the electroactivity of OTAQ and OTATQ using TBAPF6 as supporting electrolyte.
The redox behaviors of OTAQ and OTATQ were investigated using cyclic voltammetry at room temperature in a conventional three electrode cell. Electrochemical studies were carried out in ACN/DCM (5/95, v/v) solvent mixture using 0.1 M TBAPF6 as supporting electrolyte. The concentration of monomers used for polymerization was 0.003 M for OTAQ and 0.01 M for OTATQ. During the first anodic scan of OTAQ, the onset of oxidation starts at +0.46 V versus an Ag wire pseudo-reference electrode, and a single peak was observed at +0.57 V which corresponds to irreversible oxidation of the monomers (Fig. 2a). Subsequent cycling leads to a new redox couple associated with the oxidation and reduction of the polymers deposited on the ITO electrode surface. Upon repeated scanning the increasing current response of the polymer redox process illustrates that electrochemical polymerization is proceeding at the electrode surface to form an electroactive polymer film. On the anodic scans of POTAQ, polymer oxidation potential was evolved at +0.56 V and the oxidation potential increased during the repeated scanning. The reduction of the POTAQ was evolved at +0.4 V and the reduction potential decreased to 0.31 during the repeated scanning.

Repeated potential scan electrochemical polymerization of a OTAQ and b OTATQ at 100 mV/s in ACN/DCM (5:95) solution containing 0.1 M TBAPF6 on ITO
The cyclic voltammogram of OTATQ exhibits an irreversible oxidation wave at +0.75 V (Fig. 2b). Subsequent cycling leads to a new redox couple associated with the oxidation and reduction of the polymers deposited on the ITO electrode surface. POTATQ has oxidation centered at +0.91 V and the reduction peaks centered at +0.81 V. The cyclic voltammogram results showed that the oxidation potential of OTAQ is lower than that of OTATQ which was measured under the described conditions, this lower potential can be attributed to the electron withdrawing ability of the monomers. The quinoxaline unit in OTATQ is more electron-deficient and the electron withdrawing ability is stronger than OTAQ. This can be attributed to the existence of two more N atom in thiadiazole unit of OTATQ [31, 32]. So, during the electrochemical polymerization the thiophene ring in OTATQ became more electron-deficient than the thiophene ring in OTAQ and it leads to an increase in the oxidation potential of OTATQ.
The scan rate dependences of polymer films of POTAQ and POTATQ were also investigated by CV in a monomer-free electrolyte solution. A linear relationship between the peak current and scan rate demonstrates that the films were well adhered and the electrochemical processes were reversible and non-diffusion-controlled (Fig. 3).

Scan rate dependence of a POTAQ and c POTATQ in monomer free ACN/DCM (5/95) solution containing 0.1 M TBAPF6 at scan rates of 50, 100, 150, 200, 250, 300, 350, 400, 450 and 500 mV/s. Relationship of anodic (black square) and cathodic (black circled) current peaks as a function of scan rate for b POTAQ and d POTATQ film
Figure 4 presents the both p- and n-type doping properties of two polymers. The results showed that POTAQ has one peak at 0.89 V for p-type doping and one peak at 0.46 V for p-type dedoping and one peak at −1.98 V for n-type doping. The P4 has two peaks at 0.23 V and 0.87 V p-type doping and one peak at −0.24 V for p-type dedoping and one peak at −1.87 V for n-type doping. The HOMO-LUMO values were calculated by the reported method and were summarized in Table 1 [33].

Cyclic voltammogram of POTAQ and POTATQ film in 0.1 M TBAPF6/ACN/DCM (5/95,v/v) at scan rate of 100 mV/s
UV–vis-NIR analysis of polymer films
The two polymers, POTAQ and POTATQ, electrochemically deposited on the surface of ITO glass followed by washing with methylene chloride, and then the optical properties of these polymers were measured by UV-visible-NIR spectroscopy. The results (Fig. 5a) revealed that POTAQ showed two distinct absorption bands that the first band around 600–1,100 nm (λmax = 694 nm) and the second absorption band starts from 1,300 nm of which absorption peaking appears at larger than 1,600 nm. POTATQ exhibited a strong absorption bands around 700–1,600 nm and the absorption peak appeared at 1,014 nm (Fig. 5b). In Fig. 4b, there are two similar absorption bands, the first band appeared between 1,143 nm and 1,181 nm and the second band appeared between 1,337 nm and 1,484 nm. These are not real absorbance bands but the scattering peaks that were caused by the polymer particles and this phenomena are also appeared some reported works [18, 19, 21, 22, 27, 31]. These absorption bands of the two polymers can be attributed to intramolecular charge transfer between the thiophene donor and the quinoxaline acceptor unit [30].

UV–vis-NIR spectra of a POTAQ film b POTATQ film
As shown in Fig. 5b, the absorption band of POTATQ is red shifted about 320 nm compared to the first absorption band at 694 nm of POTAQ. This can be attributed to the nature of the acceptor unit of POTATQ in two aspects. Firstly, the conjugations of acceptor unit of OTATQ greater than OTAQ due to the thiadiazole ring are coplanar with the benzene ring. Secondly, the thiadiazole ring containing two N atom which has higher electron affinity and the electron withdrawing ability of acceptor unit in POTATQ, which is increased because of fusion of the thiadiazole ring onto the phenyl ring of the quinoxaline unit [31], is strong than the acceptor unit in POTAQ. The stronger electron withdrawing ability of acceptor unit leads to easier intramolecular charge transfer and cause a larger red shift of absorption peaks [32]. It’s concluded from the analysis that POTATQ has enhanced π-delocalization and intramolecular charge transfer which causes larger red shift in absorption bands.
Spectroelectrochemistry
Spectroelectrochemical analysis was performed to evaluate the optical properties of the polymers. Figure 6 shows the spectroelectrochemical series for p-doping of POTAQ between −0.4 V and +1.0 V, and for p-doping of POTATQ between −0.4 V and +1.2 V, respectively.

p-doping spectroelectrochemistry of a POTAQ film and b POTATQ film on an ITO coated glass slide in monomer free ACN/DCM (5:95) solution containing 0.1 M TBAPF6 electrolyte couple with applied potentials between −0.4 and +1.0 V for POTAQ, and between −0.4 and +1.2 V for POTATQ
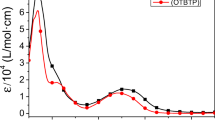
The POTAQ film is blue in color after deposited onto ITO followed by washing with methylene chloride, having two absorbance peaks centered at 694 nm and larger than 1,600 in electronic absorption spectra (Fig. 6a). The second absorption band can be attributed to intermolecular packing of POTAQ. The steric repulsion of acceptor unit and the adjacent 3-octylythiophene is lower than that between the acceptor unit and 3-octylythiophene of POTATQ, which lead to a planar geometry and increasing the intermolecular conjugation. Stepwise oxidation of the polymer film shows that the intensity of π–π* transition at 694 nm decreased and red shifted as the color changes from blue to yellow green, and then the absorption bands intensity increased during the potential changes from −0.4 V and +1.0 V, finally appear at 820 nm (red shifted 126 nm). Meanwhile, the absorption peaks in NIR region become strong. This is due to that during the electrochemical oxidation the doped extent of the polymer was increased and it leads to increase of intermolecular packing. The band gap was calculated by the peak at 694 nm as 1.06 eV.
The POTATQ film (yellow brown in color after the deposition) observed one absorption band that its absorption maxima at 1,014 nm (Fig. 6b). This absorption peak attributed to the polaronic state which is indicating the polymer film could not be dedoped to its neutral state [21]. This is because the electron withdrawing ability of acceptor unit in POTATQ is strong and the thiophene rings as donor unit become more electron-deficient. So, once the thiophene ring was oxidized, it is easy to doped. During the oxidation and reduction, injection and ejection of the counter ion become less favorable and the charges are trapped in the polymeric film. Therefore POTATQ film could not be dedoped to its neutral state. In the oxidation process, there appears a new absorption peaks which can absorb lights from 800 nm to 1,600 nm. The band gap, defined as the onset of the π–π* transition of the absorbance at 1,014 nm, was calculated as 0.78 eV. We have already discussed the reasons for greater absorption and low band gap of POTATQ film in UV–vis-NIR analysis of polymer films.
Electrochromic switching
Electrochromic switching studies for the polymers was performed to monitor the percent transmittance changes (%ΔT) as a function of time at their absorption maximum (λmax) and to determine the response time by stepping potential repeatedly between the neutral and oxidized states. During the experiment, the potential was stepwise switched between −0.4 V and +1.0 V for POTAQ and between −0.4 V and +1.2 V for POTATQ with a residence time of 10 s. In the optical contrast of POTAQ film, the changes in % transmittance between neutral (at −0.4 V) and oxidized states (at +1.0 V) were founded to be 13 % and 23 % for POTAQ as in the near-IR regions (820 nm and 1,600 nm) (Fig. 7). At the same condition, the optical contrast was determined for POTATQ as 29 % in the near-IR regions (1,500 nm). The optical contrast of the two polymer films are relatively low and it may was caused by a specific factor. It can be observe from the Fig. 6 that the absorbance at 1,600 nm was 0.18 for POTAQ and the absorbance at 1,500 nm was 0.22 for POTATQ when the applied potential was −0.4 V. The absorbance indicates that the two polymer films were not well depleted. The absorbance would reach its maxima when the polymer films p-doped by applied higher potential, the absorbance at 1,600 nm was 0.37 for POTAQ with 1.0 V and the absorbance at 1,500 nm was 0.56 for POTATQ with 1.2 V. The differences of absorbance are smaller, 0.19 for POTAQ and 0.32 for POTATQ, respectively. So, the optical contrasts of the polymer films are lower when the potential switched between the lowest and highest values.

Electrochromic switching, optical absorbance change monitored of at a 880 nm and 1,600 nm for POTAQ film and b 1,500 nm for POTATQ film in ACN/DCM (5:95) solution containing 0.1 M TBAPF6 electrolyte
The p-doping and dedoping times were calculated as 5 s for POTAQ and while the p-doping and dedoping times were determined as 10 s and 4 s for POTATQ polymer. This result indicates that the POTAQ film is easy to p-doping than POTATQ film and POTATQ film is easy to dedoping than p-doping. It is due to the difference of chemical structure of POTAQ and POTATQ. The electron acceptor unit of POTATQ has thiadiazole structure and it leads to the electron acceptor unit became more electron-deficient and has greater electron withdrawing ability than the electron acceptor unit of POTAQ. It can be attributed to the existence of two more N atom in thiadiazole unit of OTATQ [31, 32]. Therefor the charge of thiophene ring easy to transfer to quinoxaline unit and the thiophene ring were partially p-doped. So, the thiophene ring was hard to p-doping and this process needs higher applied potential than POTAQ. But the thiophene ring was easy to dedoping when applied reverse potential.
The electrochromic switching results suggest that polymer films exhibit certain electrochromic characteristics under standard conditions and can be used in celectrochromic devices.
Conclusions
In conclusion, two different electroactive donor–acceptor type monomers, in which consisting of 3-octylthiophene as the donor unit and acenaphthyl[1,2-b]quinoxaline and acenaphthyl-[1,2,5]thiadiazolo[3,4,i]quinoxaline the acceptor units, were synthesized via a Stille coupling reaction. Two novel donor–acceptor type π-conjugated polymers were synthesized by electrochemical polymerization method and investigated the influence of the changes of electron acceptor unit’s electron withdrawing ability on the optoelectronic properties of the resulting polymers. The results showed that OTAQ was polymerized in lower potential than OTATQ and spectroelectrochemical studies indicated that the band gap of POTATQ is lower than POTAQ. These differences can be attributed to the existence of two more N atoms in thiadiazole unit of OTATQ. Spectroelectrochemical experimental data showed that both POTAQ and POTATQ exhibits electrochromism via easily accessible p-type doping. Both polymer films have high π to π* electronic transitions (820 nm and 1,600 nm for POTAQ, 1,500 nm for POTATQ) and low band gaps (1.06 eV for POTAQ and 0.78 eV for POTATQ), which mainly depend on the properties of the acceptor units. POTAQ demonstrate 23 % optical contrast in NIR region with 5 s responded time and POTATQ also show 29 % optical contrast in NIR region with 10 s responded time.
References
Rougier A, Sauvet K, Sauques L (2008) Electrochromic materials from the visible to the infrared region: an example WO3. Ionics 14:99–105
Inamdar AI, Sonavane AC, Pawar SM, Kim Y, Kim JH, Patil PS, Jung W, Im H, Kim D-Y, Kim H (2011) Electrochromic and electrochemical properties of amorphous porous nickel hydroxide thin films. Appl Surf Sci 257:9606–9611
Sorar I, Pehlivan E, Niklasson GA, Granqvist CG (2013) Electrochromism of DC magnetron sputtered TiO2 thin films: role of deposition parameters. Sol Energy Mater Sol Cells 115:172–180
Liou G-S, Fang Y-K, Yen H-J (2007) Synthesis and properties of noncoplanar rigid-rod aromatic polyamides containing phenyl or naphthyl substituents. J Polym Res 14:147–155
Hsiao S-H, Guo W, Kung Y-C, Lee Y-J (2010) Redox-active and electrochromic aromatic poly(amide-imide)s with 2,4-dimethoxytriphenylamine chromophores. J Polym Res 18:1353–1364
Huang J-H, Tzuyu Huang A, Hsu C-Y, Lin J-T, Chu C-W (2012) Influence of molecular weight on silole-containing cyclopentadithiophene polymer and its impact on the electrochromic properties. Sol Energy Mater Sol Cells 98:300–307
Bicil Z, Camurlu P, Yucel B, Becer B (2013) Multichromic, ferrocene clicked poly(2,5-dithienylpyrrole)s. J Polym Res 20:228–233
Hsiao S-H, Wang H-M, Chang P-C, Kung Y-R, Lee T-M (2013) Novel organosoluble aromatic polyetheramides bearing triphenylamine moieties: synthesis, electrochemistry, and electrochromism. J Polym Res 20:154–163
Żmija J, Małachowski MJ (2011) New organic electrochromic materials and theirs applications. J Achiev Mater Manuf Eng 48:14–23
Sahin E, Sahmetlioglu E, Akhmedov IM, Tanyeli C, Toppare L (2006) Synthesis and characterization of a new soluble conducting polymer and its electrochromic devices. Org Electron 7:351–362
Chandrasekhar P, Zay BJ, McQueeney T, Birur GC, Sitaram V, Menon R, Coviello M, Elsenbaumer RL (2005) Physical, chemical, theoretical aspects of conducting polymer electrochromics in the visible, IR and microwave regions. Synth Met 155:623–627
GMURALI M, DALIMBA U, YADAV V, SRIVASTAVA R, SAFAKATH K (2013) Thiophene-based donor–acceptor conjugated polymer as potential optoelectronic and photonicmaterial. J Chem Sci 125:247–257
Shin SA, Park JB, Kim J-H, Hwang D-H (2013) Synthesis and characterization of 2,1,3-benzoselenadiazole-based conjugated polymers for organic photovoltaic cells. Synth Met 172:54–62
Tamilavan V, Song M, Agneeswari R, Kang J-W, Hwang D-H, Hyun MH (2013) Synthesis and photovoltaic properties of donor–acceptor polymers incorporating a structurally-novel pyrrole-based imide-functionalized electron acceptor moiety. Polymer 54:6125–6132
Wang T-L, Shieh Y-T, Yang C-H, Chen Y-Y, Ho T-H, Chen C-H (2013) A new low band gap donor–acceptor alternating copolymer containing dithienothiophene and fluorenone unit. J Polym Res 20:213–222
Ozyurt F, Durmus A, Gorkem Gunbas E, Toppare L (2008) A low-band gap conductive copolymer of bis-3-hexylthiophene substituted 4-tert-butylphenyl quinoxaline and 3,4-ethylenedioxythiophene. J Solid State Electrochem 14:279–283
Celebi S, Baran D, Balan A, Toppare L (2010) Enhancing electrochromic and kinetic properties of poly(2,3-bis(4-tert-butylphenyl)-5,8-di(1H-pyrrol-2-yl) quinoxaline) by copolymerization. Electrochim Acta 55:2373–2376
Pamuk M, Tirkeş S, Cihaner A, Algı F (2010) A new low-voltage-driven polymeric electrochromic. Polymer 51:62–68
Tarkuc S, Unver EK, Udum YA, Toppare L (2010) Multi-colored electrochromic polymer with enhanced optical contrast. Eur Polym J 46:2199–2205
Matsidik R, Mamtimin X, Mi HY, Nurulla I (2010) Synthesis and properties of polymer from bis-3,4-ethylenedioxythiophene substituted acenaphthenequinoxaline. J Appl Polym Sci 118:74–80
Esmer EN, Tarkuc S, Udum YA, Toppare L (2011) Near infrared electrochromic polymers based on phenazine moieties. Mater Chem Phys 131:519–524
Hellström S, Henriksson P, Kroon R, Wang E, Andersson MR (2011) Blue-to-transmissive electrochromic switching of solution processable donor–acceptor polymers. Org Electron 12:1406–1413
Sendur M, Balan A, Baran D, Toppare L (2011) Syntheses and optoelectronic properties of quinoxaline polymers: the effect of donor unit. J Polym Sci A Polym Chem 49:4065–4070
Ozdemir S, Sendur M, Oktem G, Doğan Ö, Toppare L (2012) A promising combination of benzotriazole and quinoxaline units: A new acceptor moiety toward synthesis of multipurpose donor–acceptor type polymers. J Mater Chem 22:4687–4694
Carbas BB, Kivrak A, Zora M, Önal AM (2012) Synthesis and electropolymerization of a new ion sensitive ethylenedioxy-substituted terthiophene monomer bearing a quinoxaline moiety. J Electroanal Chem 677–680:9–14
Kivrak A, Carbas BB, Zora M, Önal AM (2012) Synthesis and electropolymerization of an ion sensing and fluorescent fluorene derivative bearing a quinoxaline moiety and its analogues with different donor units. React Funct Polym 72:613–620
Tarkuc S, Udum YA, Toppare L (2012) Tailoring the optoelectronic properties of donor–acceptor–donor type π-conjugated polymers via incorporating different electron-acceptor moieties. Thin Solid Films 520:2960–2965
Wang E, Hou L, Wang Z, Hellström S, Mammo W, Zhang F, Inganäs O, Andersson MR (2010) Small band gap polymers synthesized via a modified nitration of 4,7-dibromo-2,1,3-benzothiadiazole. Org Lett 12:4470–4473
Bundgaard E, Krebs FC (2006) Low-band-gap conjugated polymers based on thiophene, benzothiadiazole, and benzobis(thiadiazole). Macromolecules 39:2823–2831
Cai T, Zhou Y, Wang E, Hellström S, Zhang F, Xu S, Inganäs O, Andersson MR (2010) Low bandgap polymers synthesized by FeCl3 oxidative polymerization. Sol Energy Mater Sol Cells 94:1275–1281
Unver EK, Tarkuc S, Baran D, Tanyeli C, Toppare L (2011) Synthesis of new donor–acceptor polymers containing thiadiazoloquinoxaline and pyrazinoquinoxaline moieties:low-band gap, high optical contrast, and almost black colored materials. Tetrahedron Lett 52:2725–2729
Yuen JD, Fan J, Seifter J, Lim B, Hufschmid R, Heeger AJ, Wudl F (2011) High performance weak donor–acceptor polymers in thin film transistors: effect of the acceptor on electronic properties, ambipolar conductivity, mobility, and thermal stability. J Am Chem Soc 133:20799–20807
Baran D, Balan A, Celebi S, Meana Esteban B, Neugebauer H, Sariciftci NS, Toppare L (2010) Processable multipurpose conjugated polymer for electrochromic and photovoltaic applications. Chem Mater 22:2978–2987
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No. 20974092, 21164011) and Xinjiang University-Institute Joint Project (No. XY110112); we appreciate the support very much.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Mahmut, M., Awut, T., Nurulla, I. et al. Synthesis of two novel acenaphthyl-quinoxaline based low-band gap polymers and its electrochromic properties. J Polym Res 21, 403 (2014). https://doi.org/10.1007/s10965-014-0403-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-014-0403-x