
Abstract
Two functional diamines, octyl-4-(3,5-diaminobenzamido)benzoate (O8, containing phenyl rings) and 3,5-diamino-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (D8, containing a phthalimide ring), were designed and successfully synthesized. Two kinds of PIs were obtained by copolymerizing 3,3′-dimethyl-4,4′-methylenediamine (DMMDA), 4,4′-oxydiphthalic anhydride (ODPA), and D8 or O8. All of the PIs possessed excellent solubility. The PI films exhibited high transmittance, and were able to align liquid crystals (LCs) vertically. In contrast to the PIs generated from O8 (PI-O8), PIs created from D8 (PI-D8) were able to align LCs vertically even after rubbing, and also exhibited better thermal stability. The temperature at which 5 % of the weight of PI-D8 was lost (T 5) was higher than that for PI-O8. The results showed that the introduction of imide groups to the side chain improved the thermal stability and rubbing resistance of PIs without sacrificing their solubility and transmittance.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aromatic polyimides (PIs) are one of the most successful classes of high-performance polymers, and have been widely used for a variety of applications, such as in electronics, composite materials, coatings, and membranes [1–4]. The popularity of PIs can be attributed to their many favorable characteristics, such as their excellent mechanical properties, long-term stability, good film-forming ability, superior chemical resistance, and the ease with which their structures can be modified.
In recent years, liquid crystal (LC) alignment layers fabricated from aromatic PIs have been the focus of intense research efforts, as they have been proposed for use in microelectronic applications that exploit the unique properties of PI materials. However, applications of conventional fully aromatic main-chain-type PIs in liquid crystal displays (LCDs) are somewhat limited by their poor processability, their brownish color, and their provision of only a small pretilt angle [1–4]. They are also insoluble and infusible as a result of high conjugation within the molecular chain and strong intramolecular and intermolecular charge-transfer complex (CTC) formation [5], which result in strong intermolecular interactions and a tendency to crystallize. Different approaches have been trialled to solve this processability issue, most of which are based on modifying their chemical structures. Flexible links such as –O–, –SO2–, –CH2–, asymmetric units, and bulky groups (hexafluoroisopropylidene, isopropylidene, etc.) are commonly employed as solubilizing moieties [6–22]. Copolymerization is also regarded as an effective method to improve the solubility of PIs by researchers. To improve the optical transparency of PIs, approaches such as introducing fluorinated groups and a twisted structure or replacing aromatic groups with alicyclic groups (forming so-called alicyclic PIs) have proven to be by far the most effective approaches [21–33]. In addition to solubility and optical transparency, the alignment materials that are applied in LCDs with large display areas, such as supertwisted-nematic-type LCDs (STN LCDs), must yield high pretilt angles. Traditional main-chain-type PI alignment layers can only provide a small pretilt angle, whereas PIs with long alkyl side chains have been found to allow high pretilt angles. However, unfortunately, the presence of side chains degrades the thermal stability of soluble PIs due to the loose chain packing and weak interchain interactions resulting from the introduction of long side chains. One of the most significant suggested ways to improve the thermal stability of soluble PIs is to adjust the chemical structure of the side chain while simultaneously achieving a high pretilt angle. However, to the best of our knowledge, few studies have focused on how to enhance the thermal stability of soluble PIs.
In the work described in the present paper, based on knowledge acquired from structure–property relationships, a new functional diamine, 3,5-diamino-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (D8), containing imide groups, was designed and synthesized. PI-D8 was then attained through the copolymerization of 4,4′-oxydiphthalic anhydride (ODPA), 3,3′-dimethyl-4,4′-methylenediamine (DMMDA), and D8 in an attempt to obtain a PI with enhanced thermal stability, high optical transparency, and a high pretilt angle, without sacrificing solubility. In order to determine the effect of the imide side chain on the thermal stability of PI, another diamine, octyl-4-(3,5-diaminobenzamido)benzoate (O8), which is similar to D8 in structure, was also synthesized. Properties such as the solubility, optical transparency, alignment ability, and—especially—the thermal stability of the PI-O8 created from O8, DMMDA, and ODPA were compared with those of PI-D8.
Experimental
Materials
4,4′-Oxydiphthalic anhydride (ODPA) was attained from the Shanghai Research Institute of Synthetic Resins (Shanghai, China), purified by recrystallization from acetic anhydride, and dried under vacuum prior to use. 3,3′-Dimethyl-4,4′-methylenediamine (DMMDA) was purchased from Shanghai EMST Corp. (Shanghai, China) and purified by recrystallization from ethanol. Isoindoline-1,3-dione, 4-nitrobenzoyl chloride, heptan-1-ol, and 1-bromoheptane was obtained from Shanghai Reagent Co. Ltd. (Shanghai, China). 3,5-Dinitrobenzoyl chloride (DNBC) was attained from Taixing Refined Chemical Co. Ltd. (Jiangsu, China) and 5 % palladium on activated carbon was obtained from Guoyao Corp. (Shanghai, China). Triethylamine (TEA) was obtained from the Chengdu Kelong Chemical Reagent Corp. (Chengdu, China). N-methyl-2-pyrrolidone (NMP) was distilled under reduced pressure after stirring over calcium hydride for 24 h and stored over 4-Å molecular sieves before use. Dimethylsulfoxide (DMSO), dimethylformamide (DMF), dimethylacetamide (DMAc), tetrahydrofuran (THF), chloroform (TCM), and m-cresol were used as received. Nematic LC E7 (n 0 = 1.521, Δn = 0.22, T n−1 = 60 °C) was purchased from Shijiazhuang Crown Chem-Tech. Co. Ltd. (Shijiazhuang, China).
Measurement
Nuclear magnetic resonance (1H NMR) spectra were obtained using a 400 MHz Varian (Palo Alto, CA, USA) Unity Inova 400 spectrophotometer using CDCl3 or DMSO as a solvent. Fourier transform infrared (FTIR) spectra were recorded with a Nicolet 560 FTIR spectrometer (Thermo Nicolet Corporation, Madison, WI, USA). A spin-coating process was executed using a KW-4A spinner from the Institute of Microelectronics of the Chinese Academy of Sciences (Beijing, China). The pretilt angles of the LCs were measured by the crystal rotation method using a pretilt angle tester from the Changchun Institute of Optics, Fine Mechanics and Physics (Changchun, China), and at least five different points on the cells were selected for measurement. The rubbing process was operated with a rubbing machine from TianLi Co. Ltd. (Chengdu, China). Thermogravimetric analysis (TGA) was performed with a DuPont (Wilmington, DE, USA) TGA2100 at a heating rate of 10 °C min−1 under a nitrogen atmosphere. Differential scanning calorimetry (DSC) testing was performed on a PerkinElmer (Waltham, MA, USA) DSC7 differential scanning calorimeter at a scanning rate of 10 °C/min under flowing nitrogen (30 cm3/min), and the T g values were read from the DSC curves at the same time. The contact angles of deionized water and methylene iodide on the surfaces of the PI films were measured using a DSA100 goniometer system (Kruss GmbH, Hamburg, Germany). Each sample was tested three times and mean contact angle as well as surface free energy values were calculated. The inherent viscosity of PI was measured at a concentration of 0.5 g dL−1 in NMP with an Ubbelohde-type viscometer at 30 °C. Gel permeation chromatography of soluble polymers was performed using an Applied Bio system (Foster City, CA, USA) at 70 °C with two PLgel 5 μm mixed-C columns (Agilent, Santa Clara, CA, USA). The energy-minimized structures of the two diamines were calculated using HyperChem v.8.0 (Hypercube, Inc., Gainesville, FL, USA).
Monomer synthesis
Synthesis of 3,5-diamino-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (D8)
The functional diamine D8 was synthesized according to Scheme 1. The synthesis is described below.

The synthetic route to the functional diamine D8
Synthesis of 5-nitroisoindoline-1,3-dione
To a 250 mL flask containing a magnetic stirrer were added isoindoline-1,3-dione (22.1 g, 0.15 mol), P2O5 (14.2 g, 0.1 mol), and 98 % H2SO4 (10.9 mL, 0.2 mol). The mixture was then added dropwise to a mixed acid (65∼68 % HNO3 12.6 mL, 0.3 mol; 98 % H2SO4 8.2 mL, 0.15 mol) in an ice-water bath. After this, the reaction mixture was warmed to room temperature for 10 h with vigorous stirring. The filter cake obtained by filtration was washed with a small quantity of ice water several times. The product was recrystallized from ethanol to afford 20.8 g (72 %) of a yellowish solid: mp 295∼297 °C.
IR (KBr, pellet), cm−1: 3,163 (N–H); 3,074 (C–H); 1,699 (C=O); 1,621, 1,463 (aromatics); 1,523, 1,337 (–NO2); 1,240 (C–N). 1H NMR (CDCl3), ppm: 10.9 (s, 1H, –NH); 9.06 (s, 1H, Ar–H); 8.64∼8.68 (d, 1H, Ar–H); 8.39∼8.35 (d, 1H, Ar–H).
Synthesis of 5-nitro-2-octylisoindoline-1,3-dione
To a 250-mL three-necked flask equipped with a condenser and magnetic stir bar were added 5-nitroisoindoline-1,3-dione (9.6 g, 0.05 mol), KOH (3.36 g, 0.06 mol), and DMF (100 mL) acting as solvent. 1-Bromoheptane (9.8 g, 0.055 mol) was added slowly after the mixture had been heated to 80 °C, and the solution was stirred for 3 h and then filtered. The filtrate was diluted with petroleum ether and transferred to a separator funnel. The organic phase was washed three times with water, separated, and dried with magnesium sulfate. The mixture was filtered, and the solvent was removed under reduced pressure to give 13.5 g (89 %) of light-yellow liquid, which was used in the next step without further purification.
IR (KBr, pellet), cm−1: 3,078 (C–H); 2,924, 2,853 (–CH2–); 1,699, 1,660 (imide); 1,621, 1,461 (aromatics); 1,525, 1,339 (–NO2); 1,232 (imide). 1H NMR (CDCl3), ppm: 9.12 (s, 1H, Ar–H); 8.90∼8.85 (d, 1H, Ar–H); 8.67∼8.60 (d, 1H, Ar–H); 3.92∼3.90 (t, 2H, –N–CH2–); 1.77∼1.69 (m, 2H, –CH2–); 1.43∼1.27 (m, 10H, –CH2–); 0.89∼0.86 (t, 3H, –CH3).
Synthesis of 5-amino-2-octylisoindoline-1,3-dione
5-Nitro-2-octylisoindoline-1,3-dione (3.04 g, 0.01 mol), THF (80 mL), and 5 % palladium on activated carbon (0.16 g) were added to a hydrogenation bottle. The bottle was secured on a Parr (Moline, IL, USA) hydrogenation apparatus, flushed three times with hydrogen, and then pressurized to 145 psi. After the mixture had been agitated at 80 °C for 8 h under a hydrogen pressure of 145 psi, it was filtered through Celite. Then the filtrate was evaporated to obtain 2.35 g (91 %) of a light-yellow liquid. Purification by flash chromatography on silica gel (petroleum ether: ethyl acetate, 5:1 v/v) afforded the pure 5-amino-2-octylisoindoline-1,3-dione in 91 % (2.35 g) yield.
IR (KBr, pellet), cm−1: 3,437, 3,357 (–NH2); 3,055 (C–H); 2,924, 2,853 (–CH2–); 1,673, 1,637 (imide); 1,575, 1,477 (aromatics); 1,526, 1,358 (–NO2); 1,241 (imide). 1H NMR (CDCl3), ppm: 8.10∼8.21 (d, 1H, Ar–H); 7.59 (s, 1H, Ar–H); 7.08∼6.99 (d, 1H, Ar–H); 3.96 (s, 2H, –NH2); 3.93∼ 3.91 (t, 2H, –N–CH2–); 1.74∼1.68 (m, 2H, –CH2–); 1.42∼1.25 (m, 10H, –CH2–); 0.89∼0.83 (t, 3H, –CH3).
Synthesis of 3,5-dinitro-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide
To a 100-mL flask placed in an ice bath were added THF (20 mL), triethylamine (3 mL), and 5-amino-2-octylisoindoline-1,3-dione (5.48 g, 0.02 mol). After the mixture had been cooled down to 0 °C, a solution of 3,5-dinitrobenzoyl chloride (7.89 g, 0.03 mol) in THF (15 mL) was added dropwise, and the mixture was maintained between 0 and 5 °C. After this, the solution was stirred for 12 h at room temperature and then filtered. The filtrate was added slowly to a vigorously stirred solution of sodium carbonate (32 g) in water (400 mL). The solid was collected by filtration and washed with distilled water three times. The product was recrystallized from a mixed solvent of ethanol/water (7/3, v/v) to afford 7.77 g (83 %) yellow crystals: mp 205∼207 °C.
IR (KBr, pellet), cm−1: 3,161 (N–H); 3,096 (C–H); 2,923, 2,853 (–CH2–); 1,699 (C=O); 1,657, 1,588 (imide); 1,540, 1,463 (aromatics); 1,539, 1,347 (–NO2); 1,236 (imide). 1H NMR (CDCl3), ppm: 9.38 (s, 1H, Ar–H); 9.27 (s, 2H, Ar–H); 8.51 (s, 1H, Ar–H); 8.11∼8.09 (d, 1H, Ar–H); 8.07∼7.99 (d, 1H, Ar–H); 3.92∼3.90 (t, 2H, –N–CH2–); 1.74∼1.68 (m, 2H, –CH2–); 1.43∼1.27 (m, 10H, –CH2–); 0.91∼0.84 (t, 3H, –CH3).
Synthesis of 3,5-diamino-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (D8)
3,5-Dinitro-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (4.68 g, 0.01 mol), THF (80 mL), and 5 % palladium on activated carbon (0.24 g) were added to a hydrogenation bottle. The bottle was secured on a Parr hydrogenation apparatus, flushed three times with hydrogen, and then pressurized to 145 psi. After the mixture had been agitated at 80 °C for 8 h under a hydrogen pressure of 145 psi, it was filtered through Celite. Tetrahydrofuran was removed by rotary evaporation under reduced pressure, and the resulting yellow product was purified by recrystallization from ethanol to give 3.66 g of white needle-shaped crystals; yield: 89.6 %.
IR (KBr, pellet), cm−1: 3,447, 3,362 (NH2); 3,163 (N–H); 3,096 (C–H); 2,925, 2,853 (–CH2–); 1,699 (C=O); 1,657, 1,588 (imide); 1,540, 1,463 (aromatics); 1,236 (imide). 1H NMR (CDCl3), ppm: 8.51 (s, 1H, Ar–H); 8.11∼8.02 (d, 1H, Ar–H); 8.07∼7.94 (d, 1H, Ar–H); 7.93 (s, 1H, –NH–); 6.62 (s, 2H, Ar–H); 5.91 (s, 1H, Ar–H); 3.97 (s, 4H, –NH2); 3.92∼3.90 (t, 2H, –N–CH2–); 1.73∼1.67 (m, 2H, –CH2–); 1.42∼1.25 (m, 10H, –CH2–); 0.87∼0.81 (t, 3H, –CH3).
Synthesis of octyl 4-(3,5-diaminobenzamido)benzoate (O8)
The functional diamine O8 was synthesized according to Scheme 2. Details of the synthesis are given below.
Synthesis of octyl 4-nitrobenzoate
To a 250-mL flask equipped with a magnetic stirrer were added heptan-1-ol (6.5 g, 0.05 mol) and triethylamine (7 mL). After the mixture had been cooled in an ice-water bath, a solution of 4-nitrobenzoyl chloride (14 g, 0.075 mol) in THF (20 mL) was added dropwise. After this, the reaction mixture was warmed to 30 °C for 12 h with stirring. The filtrate obtained from filtration was partitioned between 100 mL of ethyl acetate and 300 mL of 8 % sodium carbonate solution. The extracted organic phase was collected, washed with distilled water three times, and dried over anhydrous magnesium sulfate. The solvent was removed under reduced pressure after filtering off the inorganic solids. 12.3 g of a yellow liquid were obtained, which was used in the next step without further purification. Yield: 88 %.
IR (KBr, pellet), cm−1: 3,163 (N–H); 3,095 (C–H); 2,925, 2,855 (–CH2–); 1,700 (C=O); 1,540, 1,465 (aromatics); 1,539, 1,349 (–NO2). 1H NMR (CDCl3), ppm: 8.35 (d, 2H, Ar–H); 8.27 (d, 2H, Ar–H); 3.96∼3.91 (t, 2H, –O–CH2–); 1.74∼1.69 (m, 2H, –CH2–); 1.42∼1.25 (m, 10H, –CH2–); 0.89∼0.82 (t, 3H, –CH3).
Synthesis of octyl 4-aminobenzoate
Octyl 4-nitrobenzoate (5.58 g, 0.02 mol), THF (70 mL), and 5 % palladium on activated carbon (0.28 g) were added to a hydrogenation bottle. The bottle was secured on a Parr hydrogenation apparatus, flushed three times with hydrogen, and then pressurized to 145 psi. After the mixture had been agitated at 80 °C for 8 h under a hydrogen pressure of 145 psi, it was filtered through Celite. Tetrahydrofuran was removed by rotary evaporation under reduced pressure, and the resulting yellow product was purified by flash chromatography on silica gel (petroleum ether: ethyl acetate, 3:1 v/v) to get 4.3 g of a yellowish liquid. Yield: 86.3 %.
IR (KBr, pellet), cm−1: 3,433, 3,357 (–NH2); 3,078 (C–H); 2,924, 2,857 (–CH2–); 1,669 (C=O); 1,543, 1,467 (aromatics). 1H NMR (CDCl3), ppm: 7.73 (d, 2H, Ar–H); 6.61 (d, 2H, Ar–H); 4.24∼4.11 (t, 2H, –O–CH2–); 4.05 (s, 2H, –NH2); 1.75∼1.69 (m, 2H, –CH2–); 1.42∼1.24 (m, 10H, –CH2–); 0.91∼0.82 (t, 3H, –CH3).
Synthesis of octyl 4-(3,5-dinitrobenzamido)benzoate
To a 100-mL flask placed in an ice bath were added THF (20 mL), triethylamine (3 mL), and octyl 4-aminobenzoate (4.98 g, 0.02 mol). After the mixture had been cooled down to 0 °C, a solution of 3,5-dinitrobenzoyl chloride (7.89 g, 0.03 mol) in THF (15 mL) was added dropwise, and the mixture was maintained between 0 and 5 °C. After this, the solution was stirred for 12 h at room temperature and then filtered. The filtrate was added slowly to a vigorously stirred solution of sodium carbonate (32 g) in water (400 mL). The solid was collected by filtration and washed with distilled water three times. The product was recrystallized from a mixed solvent of ethanol/water (1/1, v/v) to afford 7.53 g (86 %) of yellow crystals: mp 201∼202 °C.
IR (KBr, pellet), cm−1: 3,161 (N–H); 2,925, 2,855 (–CH2–); 3,077 (C–H); 1,701 (C=O); 1,540, 1,465 (aromatics); 1,539, 1,347 (–NO2). 1H NMR (CDCl3), ppm: 9.40 (s, 1H, Ar–H ); 9.28 (s, 2H, Ar–H ); 8.2 (s, 1H, CO–NH ); 7.96 (d, 1H, Ar–H ); 7.75 (d, 1H, Ar–H); 4.25∼4.12 (t, 2H, –O–CH2–); 1.75∼1.69 (m, 2H, –CH2–); 1.43∼1.24 (m, 10H, –CH2–); 0.90∼0.81 (t, 3H, –CH3).
Synthesis of octyl 4-(3,5-diaminebenzamido)benzoate
Octyl 4-(3,5-dinitrobenzamido)benzoate (4.43 g, 0.01 mol), THF (70 mL), and 5 % palladium on activated carbon (0.22 g) were added to a hydrogenation bottle. The bottle was secured on a Parr hydrogenation apparatus, flushed three times with hydrogen, and then pressurized to 145 psi. After the mixture had been agitated at 80 °C for 8 h under a hydrogen pressure of 145 psi, it was filtered through Celite. Tetrahydrofuran was removed by rotary evaporation under reduced pressure, and the resulting yellow product was purified by recrystallization from ethanol to give 3.52 g of white needle-like crystals; yield: 91.8 %.
IR (KBr, pellet), cm−1: 3,431, 3,353 (–NH2); 3,163 (N–H); 3,077 (C–H); 2,923, 2,855 (–CH2–); 1,703 (C=O); 1,540, 1,467 (aromatics); 1H NMR (CDCl3), ppm: 8.01 (s, 1H, CO–NH); 7.96 (d, 1H, Ar–H); 7.75 (d, 1H, Ar–H); 6.45 (s, 2H, Ar–H); 6.11 (s, 1H, Ar–H); 4.24∼4.11 (t, 2H, –O–CH2–); 4.07 (s, 4H, 2NH2); 1.73∼1.67 (m, 2H, –CH2–); 1.43∼1.24 (m, 10H, –CH2–); 0.88∼0.80 (t, 3H, –CH3).
Polymer synthesis
A chemical imidization method was employed to obtain five kinds of PIs (see Scheme 3). The molar ratios of functional diamines (D8 or O8) to DMMDA were 0:10 (PI0), 2:8 (PI2-D8, PI2-O8), and 3:7 (PI3-D8, PI3-O8). Taking PI2-D8 as an example, NMP (35 mL), the functional diamine D8 (0.816 g, 0.002 mol), and DMMDA (1.809 g, 0.008 mol) were mixed in a 100-mL three-necked flask with magnetic stirring and blanketed in dry nitrogen. After the diamines had dissolved entirely, ODPA (3.07 g, 0.01 mol) was added, and the mixture was stirred at room temperature for 4 h to ensure the formation of poly(amide acid) (PAA). Then pyridine (1.98 g, 0.025 mol) and acetic anhydride (9.2 g, 0.09 mol) were added, and the solution was heated to 80 °C and stirred for 16 h. After cooling to room temperature, the solution was poured slowly into methanol. The polymer obtained was filtered off, washed with hot ethanol, and finally dried in a vacuum oven at 120 °C overnight. Yields were virtually quantitative in every case.
Fabrication of the liquid crystal cells
Dissolving PIs in NMP afforded solutions with a solid content of 5 %. The solutions attained were spin-coated onto clean glass substrates at a speed of 600 rpm for 15 s and 2,500 rpm for 30 s, and then heated at 120 °C for 2 h to evaporate the solvent. The PI films thus obtained were rubbed using a rubbing machine with a roller covered with rayon velvet fabric. The rubbing strength, RS, is defined as
where N is the number of times the substrates were rubbed, M (mm) is the depth of deformation of the rayon fabric due to contact with the film surface, v (mm s−1) is the velocity of the substrate stage, and n (rpm) and r (mm) are the rolling speed and radius of the roller, respectively.
The LC cells were assembled from two pieces of rubbed substrate arranged such that they had antiparallel rubbing directions, along with 20 μm thick spacers. In addition, some unrubbed substrates were used to fabricate LC cells for comparison. E7 LCs were injected into the cell gap using a capillary method at 80 °C, and the injection hole was then sealed with photosensitive epoxy glue. In this study, LC cells were preheated to 80 °C before injection to eliminate flow marks in the LC cells. The pretilt angles of LCs were measured using a crystal rotation method.
Results and discussions
Monomer synthesis
The functional diamines were synthesized according to Schemes 1 and 2.

The synthetic route to the functional diamine O8
The key step in the synthesis of 3,5-diamino-N-(2-octyl-1,3-dioxoisoindolin-5-yl)benzamide (D8) was reduction, which was carried out by H2 in a high-pressure reaction kettle in an attempt to obtain a high yield. Traditional reduction reagents such as hydrazine hydrate should be avoided, considering the hydrolysis of imide groups.
3,5-Dinitrobenzoyl chloride was utilized in the amidation step in the synthesis of each diamine (D8 and O8). Because the 3,5-dinitrobenzoyl chloride is very reactive, the reaction must be carried out at around 0 °C to prevent the generation of accessory substances. In this step, TEA acts as a catalyst that absorbs the acid produced during the reaction and speeds up the reaction.
The structures of the target diamines (D8 and O8) were confirmed by NMR (see Figs. 1 and 2) and FT-IR. All of the spectroscopic data obtained were consistent with the proposed structures.

1H NMR spectrum of the functional diamine D8 in CDCl3

1H NMR spectrum of the functional diamine O8 in CDCl3
Polymer synthesis
The commercial diamine DMMDA and dianhydride ODPA were combined with the functional diamine D8 (or O8) to attain PI-D8 bearing imide units (or PI-O8 bearing benzene units). All PIs were prepared by a two-step low-temperature polycondensation method, using acetic anhydride and pyridine as polycondensation promoters and NMP as solvent. The molecular weights achieved for these PIs were high enough to prepare creasable films by casting from polymer solutions. The results of polymerization are compiled in Table 1. From GPC measurements, it was confirmed that the polymers had high molecular weights in the range of 1.53–1.81 × 10−4 g mol−1 for M n, and the distributed indices of all side-chain-type PIs fluctuated around 1.8. The intrinsic viscosities of the PIs were also investigated, and these decreased as the number of side chains increased and correlated with the variation in the molecular weight. The two kinds of PIs had similar molecular weights and intrinsic viscosities when the content of functional diamine was kept the same. This was due to the similar structures of the two functional diamines, which present similar steric hindrances and chain packing.
The results of the elemental analysis agreed quite well with the calculated values for the proposed structures of PIs (Scheme 3). The chemical compositions of the polymers were also confirmed by IR and 1H NMR spectroscopies. As an example, the 1H NMR spectrum of PI2-D8 is shown in Fig. 3, where all of the signals can be readily ascribed to the protons of the polymer. The IR spectrum of PI2-D8 showed absorption bands that are typical of N–H stretching at 3,364 cm−1, and arise from the vibrations of amide units of side chains, as well as aliphatic C–H stretching at 2,927∼2,863 cm−1, corresponding to the methyl groups of the diamine D8 and DMMDA. Strong bands at 1,777 and 1,723 cm−1 due to the symmetrical and unsymmetrical vibrations of the two imide carbonyl groups were also observed for the novel PI, along with a sharp absorption band at around 725 cm−1, associated with skeletal vibrations of the imide rings. Absorption bands also appeared at 1,377 cm−1 and 1,270∼1,238 cm−1; these are associated with the vibrations of the C–N and C–O–C units, respectively (Fig. 4).

The synthetic route to the two PIs

1H NMR spectrum of PI2-D8

IR spectrum of PI2-D8
Solubility
The solubilities of all of the PIs were studied in different solvents (Table 2). All of the polymers were soluble in aprotic polar solvents, and even in common organic solvents such as THF and chloroform. This favorable solubility is most likely due to the steric and polar nature of the amide units of both functional diamines, which greatly favor solubility in polar organic media. Also, the steric hindrance of this –CH3 and the presence of flexible segments (–O–, –CH2–) cause the rings of the main chains to twist dramatically out of plane, resulting in a noncoplanar and contorted conformation that is capable of greatly hindering chain packing. All of these factors reduced the chain–chain interactions and enhanced solubility.
Transmittance
The UV-vis spectra of the PI films are shown in Fig. 5. The films obtained by casting from a 5 wt.% solution on a calcium fluoride (CaF2) piece showed high transmission (>90 %) in the wavelength range 400–700 nm. PI2-D8 and PI2-O8 (PI3-D8 and PI3-O8) showed equally high transmission. The good optical transparency and lighter color of the PI films resulted from decreases in intermolecular CTC interactions and electron conjugation due to the introduction of the ether units (–O–) and the side pendants. On the other hand, the introduction of an aliphatic unit (–CH2–) into the backbones of the PIs led to decreased intramolecular CTC interactions, lowering the color of these PIs [34, 35].

Transmittance of PI films
The wide-angle X-ray diffraction (XRD) curves of the PIs are shown in Fig. 6. Broad diffraction peaks are observed in all PIs, which indicates that the PIs had amorphous morphologies. Such a result explains why the films of all the PIs have good optical transmittance above 450 nm. The steric effect of side chains increases the intermolecular distance and free volume, which results in looser chain packing. Meanwhile, copolymerization can also break up the structural regularity of the PIs. Both of these factors can reduce the tendency for crystallization to occur, so amorphous morphologies were observed in this study.

The X-ray diffraction curves of the PIs
Thermal stability
Table 3 shows the thermal properties of the PIs, including the decomposition onset temperature (T d), the temperatures required to achieve weight losses of 5 % (T 5) and 10 % (T 10), and the residual weight percentage (i.e., compared to the original weight) at 800 °C (R 800). The TGA results (see Fig. 7) showed that the new PIs bearing imide side groups have excellent thermal stabilities. Compared with PI-O8, PI-D8 exhibited similar values of T d, moderately higher T 10 and R800 values, but much higher values of T 5.

TGA thermograms of the PIs
Research has shown that PIs containing side chains exhibit distinct two-step weight loss behavior [36]. The weight loss percentage during the first degradation was equivalent to the weight fraction of the side chain, indicating that the values of T d and T 5 are closely related to the structure of the side chain. Both of the functional diamines possessed the same long alkyl chains, and the backbones of the PIs were also the same, so similar values of T d were observed. However, the introduction of phthalimide into the side chains of PI-D8 increased both the rigidity of D8 and the weight ratio of side chains, resulting in higher values of T 5 than that of PI-O8. The second degradation corresponded to the thermal degradation of the polymer main chain, so similar values of T 10 and R800 were discovered for both PIs.
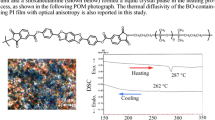
Thermal phase transitions of the PI samples were examined by performing DSC in nitrogen. The glass transition temperatures (T g) of the PIs are listed in Table 3, and the DSC curves are shown in Fig. 8. The two kinds of PIs exhibited parallel thermal phase transitions. For example, the values of T g for PI3-D8 and PI3-O8 were almost the same. The similar spatial configurations of the two functional diamines designed in this work resulted in similar intermolecular interactions, chain packing, and free volumes in the two polymer systems, so similar T g values were obtained for both.

The DSC curves of the PIs
Alignment ability and pretilt angle of the liquid crystal
The pretilt angle is an important parameter that determines the electro-optical properties of LCD devices. The pretilt angle is affected by various factors, such as surface tension, surface morphology, steric effects, and electronic interactions of the LC with the alignment layer. It may therefore be influenced by the processing conditions, such as a rubbing process, as well as the chemical structure of the PI. As is well known, rubbing is widely used to realize homogeneous alignment of the LC molecules on the polymer surface [37]. In this work, the effect of rubbing on the pretilt angles of LC cells was also investigated.
As shown in Table 4, the PIs containing side chains induced a pretilt angle of 90° before the rubbing process and uniform alignment of the LCs. However, after the rubbing process, PIs with different functional diamines induced different LC pretilt angles. PI2,3-D8 was still able to align the LCs vertically, and the pretilt angles of PI2,3-D8 stayed above 89° after the rubbing treatment, which were higher than those of PI2,3-O8. The energy-minimized structures of the functional diamines D8 and O8 were calculated via HyperChem v.8.0. As shown in Scheme 4, the torsional angle between the two terminal atoms of a four-atom strand (C1, C10, C11, C12) in O8 is about 62.8°, while the torsional angle between the two terminal atoms of a four-atom strand (C1, C8, C9, C10) in D8 is about 93.4°, which is much larger than that of O8. Further, D8 has a more rigid connecting group (a phthalimide ring) than O8 (a phenyl ring), which may restrict the movement of the long side chain into the polymer surface, resulting in low surface energy and enrichment of nonpolar alkyl side chains in the outmost layer of the PI surface. When the rubbing process was performed, the side chain layer of D8 was able to withstand the rubbing strength of the cloth and ensure a stable arrangement of side chains. The interactions of the alkyl side chains and LCs resulted in higher pretilt angles for rubbed PI2,3-D8 than for PI2,3-O8.

The energy-minimized structures of the two functional diamines D8 and O8 (calculated via HyperChem v.8.0)
The contact angles of deionized water and methylene iodide on the surfaces of the PI films were measured, and the calculated surface free energies are listed in Table 4. The surface free energies of PI-D8 and PI-O8 are quite similar before rubbing because they both have the same alkyl side chain. After rubbing (RS = 200 mm), the surface energy of PI-O8 increases more than that of PI-D8. This difference may be due to the increase in side-chain rigidity. Because of the increased rigidity, the nonpolar alkyl endgroups in the side chain could still orient out of the film plane and mask the polar groups in the backbone after rubbing. Hence, no obvious change in surface energy was observed on the surface of PI-D8.
Conoscopic observations of the LC cells were performed with a polarized optical microscope. As shown in Fig. 9, a dark crossed brush was clearly seen that did not move when the LC cell was rotated before and after rubbing. This result further proves that the vertical alignment was induced by PI with phthalimide side chains.

Polarized optical microscopic images of a vertical LC. All of the images were captured under crossed polarizers. The conoscopic images are shown in the corner of the picture. a Before rubbing; b after rubbing. The alignment film is PI2-D8
Conclusions
A series of PIs were synthesized from ODPA, DMMDA, and a functional diamine (D8 or O8) that had a long nonpolar alkyl side group connected with a rigid imide ring or benzene ring. The structures of the intermediates, diamines, and PIs were confirmed by analyzing their FT-IR and 1H NMR spectra. All of the PIs obtained were soluble in polar aprotic solvents and low-boiling-point solvents. Films of the PIs showed favorably high transmittances of >90 %. The thermal stability of PI-D8 with phthalimide side chains was better than that of PI-O8 with side chains containing phenyl units. The temperature T 5 of PI-D8 was much higher than that of PI-O8. Meanwhile, films of PI-D8 were still able to align the LCs vertically after rubbing, and showed better rubbing resistance than those of PI-O8. Hence, the introduction of rigid imide units into the side chains improved the thermal stability of PI and its rubbing resistance without sacrificing its solubility and transmittance. This kind of PI could be a suitable candidate for alignment layers used in the manufacture of high-performance vertical alignment mode LCDs.
References
Sroog CE (1976) Polyimide. J Polym Sci Macromol Rev 11:161–208
Guo C-J, Sun Z, Xia S-L, Wang Y-H (2012) A preliminary inquiry into the conformation of side chains on polyimide film and the mechanism of the vertical alignment of liquid crystals induced by polyimides with side chains. Liq Cryst 39:721–728
Thiruvasagam P (2012) Synthesis of unsymmetrical diamine and polyimides: structure–property relationship and applications of polyimides. J Polym Res 19:9965–9972
Cosutchi AI, Hulubei C, Stoica I, Ioan S (2011) A new approach for patterning epiclon-based polyimide precursor films using a lyotropic liquid crystal template. J Polym Res 18:2389–2402
Hasegawa M, Mita I, Kochi M, Yokota R (1989) Charge-transfer emission spectra of aromatic polyimides. J Polym Sci C Polym Lett 27:263–269
Li Y, Wang Z, Li G, Ding M, Yan J (2012) Synthesis and properties of polyimides based on isomeric (4,4′-methylenediphenoxyl) bis(phthalic anhydride)s (BPFDAs). J Polym Res 19:9772–9778
Yi L-F, Xia S-L, Sun Z, Liu M-S, Wang Y-H (2012) A study of the stabilization of vertical alignment for liquid crystals by increasing the side-chain rigidity of polyimides. Polym Int 62:658–664
Cheng S-H, Hsiao S-H, Su T-H, Liou G-S (2005) Novel aromatic poly(amine-imide)s bearing a pendent triphenylamine group: synthesis, thermal, photophysical, electrochemical and electrochromic characteristics. Macromolecules 38:307–316
Huang X, Huang W, Fu L, Yan D (2012) Synthesis and characterization of thioether-containing polyimides with high refractive indices. J Polym Res 19:9790–9794
Fang Y-Q, Wang J, Zhang Q, Zeng Y, Wang Y-H (2010) Synthesis of soluble polyimides for vertical alignment of liquid crystal via one-step method. Eur Polym J 46:1163–1167
Huang B, Sun Y, Wang G, Cai M (2013) Synthesis and properties of novel random copolymers of poly(ether ketone ether ketone ketone)-poly(ether ketone imide). J Polym Res 20:81–90
Anannarukan W, Tantayanon S, Zhang D, Alenman EA, Modarelli DA, Harris FW (2006) Soluble polyimides containing trans-dianminotetraphenylporphyrin: synthesis and photoinduced electron transfer. Polymer 47:4936–4945
Liou G-S, Hsiao S-H (2002) Synthesis and properties of new soluble triphenylamine-based aromatic poly(amine amide)s derived from N,N′-bis(4-carboxyphenyl)-N,N′-diphenyl-1,4-phenylenediamine. J Polym Sci A Polym Chem 41:94–105
Hsiao S-H, Chang Y-M, Chen H-W, Liou G-S (2006) Novel aromatic polyamides and polyimides functionalized with 4-tert-butytriphenylamine groups. J Polym Sci A Polym Chem 44:4579–4592
Liou G-S, Yang Y-L, Su YO (2006) Synthesis and evaluation of photoluminescent and electrochemical properties of new aromatic polyamides and polyimides with a kink 1,2-phenylenediamine moiety. J Polym Sci A Polym Chem 44:2587–2603
Behniafar H, Boland P (2010) Heat stable and organosoluble polyimides containing laterally-attached phenoxy phenylene groups. J Polym Res 17:511–518
Yang C-P, Su Y-Y (2006) Novel organosoluble and colorless poly(ether imide)s based on 3,3′-bis[4-(3,4-dicarboxyphenoxy)phenyl]phthalide dianhydride and aromatic bis(ether amine)s bearing pendent trifluoromethyl groups. J Polym Sci A Polym Chem 44:3140–3152
Nasab SMA, Ghaemy M (2011) Synthesis and characterization of new polyamides and polyimides containing dioxypyrimidine moiety in the main chain with bulky imidazole pendant group: solubility, thermal and photophysical properties. J Polym Res 18:1575–1586
Li F, Ge JJ, Honigfort PS, Fang S, Chen J-C, Harris FW, Cheng SZD (1999) Dianhydride architectural effects on the relaxation behaviors and thermal and optical properties of organo-soluble aromatic polyimide films. Polymer 40:4987–5002
Li F, Fang S, Ge JJ, Honigfort PS, Chen JC, Harris FW, Cheng SZD (1999) Diamine architecture effects on glass transitions, relaxation processes and other material properties in organo-soluble aromatic polyimide films. Polymer 40:4571–4583
Faghihi K, Hajibeygi M, Shabanian M (2010) New photosensitive and optically active organo-soluble poly(amide-imide)s from N,N-(bicycle[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids and 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one: synthesis and characterization. J Polym Res 17:379–390
Liaw D-J, Chang F-C (2004) Highly organosoluble and flexible polyimides with color lightness and transparency based on 2,2-bis[4-(2-trifluoromethyl-4-aminophenoxy)-3,5-dimethylphenyl]propane. J Polym Sci A Polym Chem 42:5766–5774
Kim Y-H, Kim H-S, Kwon S-K (2005) Synthesis and characterization of highly soluble and oxygen permeable new polyimides based on twisted biphenyl dianhydride and spirobifluorene diamine. Macromolecules 38:7950–7956
Ichino T, Sasaki S, Matsuura T, Nishi S (1990) Synthesis and properties of new polyimides containing fluorinated alkoxy side chain. J Polym Sci A Polym Chem 28:323–331
Matsuura T, Hasuda Y, Nishi S, Yamada N (1991) Polyimide derived from 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl. 1. Synthesis and characterization of polyimides prepared with 2,2′-bis(3,4-diacarboxyphenyl)hexafluoropropane dianhidride or pyromellitic dianhydride. Macromolecules 24:5001–5005
Matsuura T, Ishizawa M, Hasuda Y (1992) Polyimides derived from 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl. 2. Synthesis and characterizationz of polyimides prepared from fluorinated benzenetetracarboxylic dianhydrides. Macromolecules 25:3540–3545
Matsuura T, Ando S, Sasaki S, Yamamoto F (1994) Polyimides derived from 2,2′-bis(trifluoromethyl)-4,4′-diaminobiphenyl. 4. Optical properties of fluorinated polyimides for optoelectronic components. Macromolecules 27:6665–6670
Matsumoto T, Kurosaki T (1997) Soluble and colorless polyimides from bicycle[2.2.2]octane-2,3,5,6-tetracarboxylic 2,3:5,6-dianhydrides. Macromolecules 30:993–1000
Matsumoto T, Feger C (1998) Optical properties of polyalicyclic polyimides. J Photopolym Sci Technol 11:231–236
Volksen W, Cha HJ, Sanchez MI, Yoon DY (1996) Polyimides derived from nonaromatic monomers: synthesis, characterization and potential applications. React Funct Polym 30:61–69
Liu J-G, Nakamura Y, Suzuki Y, Shibasaki Y, Ando S, Ueda M (2007) Highly refractive and transparent polyimides derived from 4,4-[m-sulfonylbis(phenylenesulfanyl)]diphthalic anhydride and various sulfur-containing aromatic diamines. Macromolecules 40:7902–7909
Liu JG, He MH, Zhou HW, Qian ZG, Wang FS, Yang SY (2002) Organosoluble and transparent polyimides derived from alicyclic dianhydride and aromatic diamines. J Polym Sci A Polym Chem 40:110–119
Choi M-C, Wakita J, Ha C-S, Ando S (2009) Highly transparent and refractive polyimides with controlled molecular structure by chlorine side groups. Macromolecules 42:5112–5120
Yang C-P, Chen R-S, Wei C-S (2002) Synthesis and properties of soluble and light-colored poly(amide-imide-imide)s based on tetraimide-dicarboxylic acid condensed from 4,4-oxydiphthalic anhydride, 2,2-bis[4-(4-aminophenoxy)phenyl]sulfone, and m-aminobezoic acid, and various aromatic diamines. J Polym Res 9:97–105
Wang L, Chang P, Cheng C-L (2006) Structural effects of pendant groups on thermal and electrical properties of polyimides. J Appl Polym Sci 100:4672–4678
Kim SI, Ree M, Shin TJ, Jung JC (1999) Synthesis of new aromatic polyimides with various side chains containing a biphenyl mesogen unit and their abilities to control liquid-crystal alignments on the rubbed surface. J Polym Sci A Polym Chem 37:2909–2921
Lee YJ, Choi JG, Song I-K, Oh JM, Yi MH (2006) Effect of side chain structure of polyimides on a pretilt angle of liquid crystal cells. Polymer 47:1555–1562
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant no. 51173115), the Ministry of Education (the Foundation for Ph.D. training, grant no. 20110181110030) of China, and the Scientific Research Foundation for Returned Overseas Chinese Scholars, State Education Ministry, and Science (no. 20071108-18-12).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Xia, S., Yi, L., Sun, Z. et al. The effect of phthalimide side chains on the thermal stability and rubbing resistance of polyimide used as a liquid crystal vertical alignment layer. J Polym Res 20, 219 (2013). https://doi.org/10.1007/s10965-013-0219-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-013-0219-0