
Abstract
The dispersion of UV-curable waterborne polyurethane acrylate possessing perfluorooctanoate side-chains (FPUA) terminated with pentaerythritol triacrylate (PETA) was synthesized by incorporating fluorine groups into the side chains of the polyurethane macromolecular from condensation polymerization by using perfluorooctanoate glycerate (OF-diols) as chain extender, while the polyurethane macromolecular was synthesized from isophoronediisocyanate (IPDI), dimethylol propionic acid (DMPA), and poly (propylene glycol) (PPG) etc. The chain extender was prepared from 2,2-dimethyl-1,3-dioxolane-4-methyl perfluorooctanoate (OF-diols-pg) by acid hydrolysis with trifluoroacetic acid. The structure of FPUA was characterized by FTIR, NMR, elemental analysis, UV–vis and Mass spectra, and the properties of the synthesized FPUA emulsion were also discussed. The effect of fluorine content on the UV-curing dynamics was studied and the UV-curing speed decreased with increment in fluorine content. Contact angle measurements shown that the hydrophobic property of the surface greatly increased by incorporation of fluorine into the polyurethane chain. In addition, scanning electron spectroscopy (SEM) analysis was applied to characterize the morphology of the fracture surface of the UV-cured FPUA which became phase-mixed after annealing.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Waterborne coatings have been widely investigated due to industrial demand according to intensifying environmental regulations [1–3]. Meanwhile, polyurethanes have been popularly employed as principal resin for conventional waterborne coatings for their prominent performance. As for aqueous polyurethane, ionic and/or nonionic hydrophilic segments should be incorporated into polyurethane molecular structure such as carboxyl or poly(ethylene glycol) (PEG) groups [4, 5]. The hydrophilic segments act as internal emulsifier, while polyurethane forms dispersion in water without strong external shear force or additional surfactant [6]. However, hydrophilic segments of aqueous polyurethane adversely affect water and soil repellency due to the relatively high surface free energy [7]. One method to complement the defect is incorporation of strong hydrophobic segments in the polyurethane structure [8, 9]. Recently, a number of work has been done on the synthesis and characterization of waterborne polyurethane by incorporating fluorinated polymers with regard to the high attractive bulk and surface properties, e.g., excellent environmental and thermal stability, water and oil repellency, biocompatibility, chemical resistance and inherently low interfacial free energy [10–14]. Ashish et al. had synthesized the segmented polyurethane (SPU) by reacting hydroxyl (polyleneoxy)-propylether-terminated PDMS block copolymer (HO-PEG-PDMS-PEG-OH) with 4,4'-methylene-bis(phenylene isocyanate) (MDI) and then fluorine segment was incorporated by following reaction with 1,2-diol functional PFPE and fluorinated chain extension [15]. Similar work also had been reported by Xu et al. [16] and Damien et al. [17].
Furthermore, UV-curable waterborne coatings have received increasing attentions in the past decade because of their advantages of environmental protection, lower energy consumption, high curing speed, rheological controlling, and adaptation to spraying [18–23]. Kim had reported a method to modify the surface of UV cured polyurethane by blending the fluorinated PU with base waterborne PUA prior to dispersion in water [24]. However, little work has been done to prepare UV-curing waterborne fluorinated polyurethane from condensation polymerization. In this study, different amounts of fluorine-containing UV-curable waterborne polyurethane acrylate possessing perfluorooctanoate side-chains (FPUA) was synthesized by incorporation the fluorinated chain extender into the hard segment of the polyurethane by means of condensation polymerization. The synthesis procedure, curing dynamics, surface properties, and morphology of the UV-cured FPUA film will be described.
Experimental part
Materials
Perfluorooctanoic acid was purchased from 3 F New Materials Co. Ltd. (90% purity). Isophoronediisocyanate (IPDI) was purchased from the HÜls Co. Dimethylol propionic acid (DMPA, Huangshan Dunxi Plastic Factory) and poly (propylene glycol) (PPG, Mn = 1000 g/mol) were dried overnight in vacuum before use. Pentaerythritol triacrylate (PETA) was provided by Sartomer Company Inc. 2-Hydroxy-4-(2-hydroxyethoxy)-2-methylpropiophenone (Irgacure 2959, Ciba-Geigy Co.) was used without further purification. Triethylamine (TEA, Guangzhou Chemical Factory) was used as neutralization reagent. N-methyl-2-pyrrolidone (NMP), dimethyl fomamide (DMF) and acetone were purchased from commercial sources and stored over 4 Å molecular sieve. Thionyl chloride (SOCl2), stannous caprylate (SC), 2,2-dimethyl-1,3-dioxolane-4-methanol (97% purity), trifluoroacetic acid, tetrahydrofuran (THF) purchased from commercial source were used as received.
Synthesis of perfluorooctanoate glycerate (OF-diols)
The fluorinated chain extender, i.e. perfluorooctanoate glycerate (OF-diols), was prepared from 2,2-dimethyl-1,3-dioxolane-4-methyl perfluorooctanoate (OF-diols-pg) by acid hydrolysis. The typical experimental procedure was illustrated in Scheme 1. Perfluorooctanoic acid (10 g, 0.0242 mol) were dissolved in SOCl2 (5.28 ml, 0.0726 mol) and stirred for 4 h at 70 °C under refluxing with one drop DMF as catalyst. The mixture solution was standing to get the colorless solution, and then the excessive SOCl2 was removed by vacuum distillation. Perfluorooctanoyl chloride (PFOC) was obtained as a colorless liquid. Afterward, 2,2-dimethyl-1,3-dioxolane-4-methanol (2.2 g, 0.0167 mol), TEA (2.4 ml) and THF (20 ml) were mixed in a round flask. Stoichiometric PFOC dissolved in THF was dropped slowly into the flask under ice bath and stirred for another 24 h. Finally the mixture was filtered and the solvent was evaporated by rotary evaporation. The crude OF-diols-pg was purified by silica gel column chromatography (petroleum ether/ethyl acetate 20:1) to yield 8.8 g of OF-diols-pg (69%). OF-diols-pg was obtained as a colorless liquid (at room temperature). IR (KBr, cm−1): 2990.3 (CH3), 1784.5 (C = O), 1240.2, 1207.9, 1147.3 (CF), 1083.7, 1059.5 and 1015.2 (C-O-C). 1H NMR (300 MHZ, CDCl3, δ in ppm), (Fig. 1a): 1.36 (s, 3H), 1.42 (s, 3H), 3.78–3.83 (m, 1H), 4.08–4.11 (m, 1H), 4.37–4.42 (m, 3H).

Synthesis route of OF-diols

1H NMR spectra of OF-diols-pg (a) and OF-diols (b)
Trifluoroacetic acid (6 ml) and OF-diols-pg (4.06 g, 0.0076 mol) were dissolved in 60 ml CH2Cl2 under ice bath. Then the solution was stirred for 24 h. After that the solvent was evaporated by rotary evaporation. The crude product was purified by silica gel column chromatography (petroleum ether/ethyl acetate 2:1) to yield 2.0 g of OF-diols (56%). OF-diols was obtained as a white solid (at room temperature). M.p. 27–28 °C. MS: m/z (%): 489.02 (M + H)+. Anal. Calcd for C11H7F15O4 (488.01): C, 27.07; H, 1.45; F, 58.38. Found: C, 27.09; H, 1.44; F, 58.39. IR (KBr, cm−1): 3356.6 (OH), 2930.3 (CH2), 1772.7 (C = O), 1211.9, 1147.3 (CF); 1H NMR (300 MHZ, CDCl3, δ in ppm), (Fig. 1b): 1.95 (s, 3H), 3.64–3.68 (m, 1H), 3.75–3.80 (m, 1H), 4.04–4.07 (m, 1H), 4.44–4.51 (m, 2H).
Synthesis of UV-curable fluorinated polyurethane acrylate (FPUA)
A series of waterborne FPUA were synthesized by three-step processes according to the reaction scheme (Scheme 2) and the feed ratios were shown in Table 1. In the beginning, PPG and DMPA were added into a three-neck flask equipped with a stirrer, thermometer, and reflux condenser under N2 atmosphere at 60 °C. IPDI containing 0.1 wt.% SC was dropped into the reactor and the mixture was heated to 85 °C for 5 h before chain extender OF-diols were added into the reaction. Then the mixture was stirred for another 7 h until the -NCO content reached the theoretical value. Acetone was added to adjust the viscosity during the reaction. After that the pre-polymer was cooled to 50 °C, and then PETA were added and reacted for 24 h until the −NCO disappeared in the FTIR. TEA was added into the solution to neutralize at room temperature for 2 h and the product was allowed to disperse into distilled water with vigorous stirring (1200 r/pm). The UV-curable waterborne FPUA was obtained after removal of the acetone from the emulsion by rotary vacuum evaporation.

Schematic diagram for the synthesis of UV-curable FPUA emulsion
Preparation of UV-cured films
The UV-cured FPUA films were prepared by casting the aqueous dispersion containing 4 wt.% photoinitiator Irgacure 2959 based on the ionomer onto a glass plate and then by curing under a medium pressure mercury lamp (1000 W) at room temperature.
Instrumentation
Fourier transform infrared (FTIR) spectra was recorded by using a NICOLET MX-1E FTIR instrument. 1H NMR spectra was recorded by using a BRUCK AMX300 NMR instrument (300 MHz). 19 F NMR data was obtained with BRUCK AC-P (300 MHz), and deuterodimethyl sulfoxide was used as deuterated solvent. UV–vis spectra was measured on Shimadzu UV-2401 spectrophotometer. Melting points were determined with an Electrothermal 9100 apparatus. Mass spectra were recorded on a Finnigan-Matt 8430 mass spectrometer operating at an ionization potential of 70 eV. Elemental analyses for C, H, and F were performed using a Heraeus CHNO-Rapid analyser. The particle size of the FPUA dispersions was measured on a particle analyzer Autosizer Loc (Malvern) by laser light-scattering method. Zeta potential (ζ) of the emulsion was measured with Zetasizer Nano ZS surface potential particle size analyzer. Differential scanning calorimetry (DSC) model 200 C (Seiko) was used to examine the thermal properties of FPUA at temperatures ranging from −80 to 100 °C and 10 °C/min of heating rate. The curing kinetics of the UV-curable FPUA was investigated by gel content method [4]. And the gel content was determined on the cured films by measuring the weight loss after 24 h extraction with acetone at room temperature. The weights of cured film before (W0) and after extraction (W) were measured to calculate the gel content according to:
Contact angles were measured with an OCA 20 (Data Physics) contact angle goniometer on 3 μl of distilled water at 20 °C, and the result reported was the mean value of 3 replicates. The SEM measurement was performed on FEI scanning electron microscope (Model INSPECTF) while the UV-cured FPUA films were split by liquefied nitrogen.
Results and discussion
Synthesis of perfluorooctanoate glycerate (OF-diols)
OF-diols was prepared from 2,2-dimethyl-1,3-dioxolane-4-methyl perfluorooctanoate (OF-diols-pg) by acid hydrolysis with trifluoroacetic acid, and the chemical structure was determined by 1H NMR analyses. 1H NMR spectra of OF-diols-pg (a) and OF-diols (b) are shown in Fig. 1, respectively. There are two methyl groups (δ = 1.36–1.42 ppm) in OF-diols-pg, as well as the peak of methylene protons bonded with oxygen at δ = 3.78–4.11 ppm and the peak at δ = 4.37–4.42 ppm corresponding to O = C-OCH2-CHO- group (Fig. 1a) proved the structure of the OF-diols-pg. Meanwhile, the disappearance of methyl signals (1.36–1.42 ppm) from OF-diols-pg and the appearance of hydroxyl signals (1.95 ppm) in OF-diols indicated the reaction of acid hydrolysis from OF-diols-pg. In addition, other protons of OF-diols were observed in the corresponding area indicated in Fig. 1b, e.g., methine signal at δ = 3.64–3.80 ppm and −CH2OH signal at δ = 4.04–4.07 ppm.
Synthesis and characterization of UV-curable waterborne FPUA emulsions
A series of UV-curable waterborne FPUA with various amount of OF-diols was prepared by three-step reaction as shown in Scheme 2. By variation the ratio of DMPA/OF-diols, the content of fluorine introduced into the hard segment can be controlled. As listed in Table 1, the fluorine weight content was ranging from 5% to 15% while COOH content was varying from 3.24% to 2.06%, respectively.
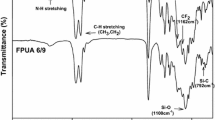
FTIR spectra was used to follow the reaction steps of the synthesis of UV-curable FPUA emulsion (Fig. 2). First, Isophoronediisocyanate reacted with DMPA and PPG to form isocyanato-terminated polyurethane pre-polymer (PU) with pendant carboxyl group. The formation of PU was evidenced by the emergence of the strong absorption at 1529 cm−1 (N-H), 1715 cm−1 (C = O), 3415 cm−1 (N-H), and absorption of −NCO at approximately 2262 cm−1(Fig. 2a). And the formation of chain extension from OF-diols (FPU) was confirmed by the substantial decrease absorption of −NCO at 2262 cm−1 and the stretching vibration of CF observed at ~1173 cm−1 in Fig. 2b. In addition, 19 F NMR for purified FPU showing the characteristic peaks remarkably at ~80 ppm (−CF3), ~111 ppm (−CF2-CH2), ~121 ppm and ~125 ppm (−CF2-CF2-) intensified the structure of CF2 and CF3 (Fig. 3). The completion of end capping reactions to form FPUA had been confirmed by the disappearance of −NCO absorption peak at 2262 cm−1, shown in Fig. 2c. However, the stretching vibration of C = C was not observed at 1650 cm−1, which may be covered by the strong stretching vibration of carbonyl bond. Furthermore, the maximum absorption wavelength (λmax) of the purified FPUA at 275 nm measured by UV–vis analysis in Fig. 4 proved the existence of conjugated double bond.

FTIR spectra of the three reaction steps synthesizing UV-curable FPUA emulsion PU (a), FPU (b) and FPUA (c)

19F NMR spectra of FPU

UV–vis analysis of UV-curable FPUA film
The basic characterization of the UV-curable waterborne FPUA emulsion was summarized in Table 1. The volume-average particle size (Dv) only had a slightly change with the variation of fluorine content and varied from 109 nm to 125 nm. The particle size distribution shown a good normal distribution and the distribution index (PDI) was around 0.25, which indicated that the particle size was less sensitive to fluorine content in this waterborne UV-curable system. This may because PETA was used as end capping reagent while the fluorinated segments formed the core of the particle, and the same result was also reported by Jia Bing Dai et al. [12] and Jiang Min et al. [25]. Moreover, because of incorporating anion COO− into the backbone chain of the polyurethane, the particles had a strong negative surface charge confirmed by Zeta potential. The value of the surface charge density of the synthesized fluorinated polyurethane acrylate was all below −27 mV, which meant the obtained emulsion had a good mechanical stability.
Kinetics of the UV-curing process
The gel content was measured by Soxhlet extraction to estimate the extent of curing. Figure 5 shows the change in gel content as a function of UV-curing time for the FPUA consisting of different fluorine content. And the rate of UV-curing process was determined by the slope of the gel content versus time plots. The polymerization rate increased rapidly at the beginning of the irradiation, then dropped in the later curing process which indicated one of the most characteristic features, i.e. auto-acceleration phenomenon. This may be due to extremely restricted diffusion of radicals in the highly cross-linked polymer and make termination more difficult [26]. Moreover, the final gel content was in the order of FPUA1>FPUA2>FPUA3 and varied from 95% to 77%. This suggested that the UV-curing speed decreased by the increment of fluorine content, which meant that the fluorine chain had a negative effect on the UV-curing speed of the fluorinated polyurethane acrylate. Bai et al. also found that the introduction of hydrophobic silica chain into the polyurethane system had a bad effect on the UV-curing kinetic [27].

The relationship between the curing time and gel content
Evaluation of contact angles
The hydrophobic property of UV-cured FPUA film surface was investigated, and the relationship between the fluorine content and contact angle is shown in Fig. 6. However, the contact angle of the UV-cured FPUA was small and the value of contact angle was nearly the same with the UV-cured polyurethane acrylate which did not contain fluorine. This may be because of fluorine chain needed a thermodynamic driving force for migration to the surface [28]. In addition, the Tg of hard segment measured by DSC before UV-cured was about 40 °C and above the room temperature. Therefore, during the UV-curing process, the fluorine chain attached to the hard segment could not migrate to the surface at room temperature. Moreover, Kim found that the contact angle of the waterborne fluorinated polyurethane film could increase when the film annealed at high temperature [6], which was also supported by our experiments, the contact angle of UV-cured FPUA films increased while the films were annealed at 120 °C for 12 h. Apparently, as shown in Fig. 6, the contact angle increased sharply with the increment of fluorine content, and the surface of the film become hydrophobic when the fluorine content was above 12.3 wt.%, which further proved that the segment which had strong hydrophobic perfluoroalkyl side chains migrated toward the surface through annealing and rendered the surface more hydrophobic [8].

The relationship between the fluorine content and contact angle after the FPUA film was annealed at 120 °C for 12 h
Characterization of bulk morphology
For confirming the morphology of the UV-cured FPUA, fracture surface was examined by scanning electron spectroscopy (SEM) [14, 29], as shown in Fig. 7. Because of the intrinsic higher incompatibility between fluorinated side chains, soft segment and hard segment in the polyurethane chains for large differences in chemical structure, there are phase separations in this materials. With the increasing content of fluorine, fracture surface morphology would transform from an equably size distribution of ball-like dispersion phase (fluorine content of 5 wt.%), i.e. sea-island structure, to a slight rough surface with disappearance of sea-island structure (fluorine content of 9.5 wt.%), and then to a much rough surface with a maldistribution of bigger ball-like dispersion phase (fluorine content of 12.3 wt.%). Finally, a much great micro-phase separation, as well as small cavities appeared while fluorine content reached 15 wt.%. The macrostructure of FPUA1 was homogeneous, while the SEM results indicate the begining of micro-phase separation. We deem that the dimension of the balls attributes to the different fluorine content in the FPUA films. However, with the increasing fluorine content from 5 wt.% to 9.5 wt.%, the size of balls tends to decreased, which means phase separation decrease. And then the phase separation showed a further increase while fluorine content varying from 9.5 wt.% to 15 wt.%. Meanwhile, the morphology of the UV-cured FPUA films was also investigated after it were annealed at 120 °C for 12 h by SEM (Fig. 8), which indicated that the annealed UV-cured FPUA films had a worse phase separated structure than that of the films without anneal. These results shown that the fracture surface presented three domains, i.e., the surface attached to the glass surface (I), the surface to the air (III) and the medium layer (II) (Fig. 8). Furthermore, the thickness of the surface to the air increased by the increment of fluorine content, which probably due to the more fluorine chain migrated to the surface with thermodynamic driving force through anneal. However, the sample with the fluorine content of 9.5 wt.% was an exception, which shown a uniform and homogeneous smooth fracture surface (Fig. 8b). And it was in accordance with the tendency of the morphology before anneal, which may be because the fluorine chain, soft segment and hard segment of polyurethane chain were miscible and phase separation was not obvious at the fluorine content of 9.5 wt.%.

SEM micrograph of fracture surface for the UV-cured FPUA before anneal a FPUA1, b FPUA2, c FPUA3, d FPUA4

SEM micrograph of fracture surface for the UV-cured FPUA after anneal a FPUA1, b FPUA2, c FPUA3, d FPUA4
Conclusions
A series of UV-curable FPUA dispersions has been synthesized by introducing OF-diols into the side chain of polyurethane by means of condensation polymerization. The particle size of the emulsion had a slightly change with the variation of fluorine content and varied from 109 nm to 125 nm. Meanwhile, the particle size distribution shown a good normal distribution and the distribution index (PDI) was around 0.25. The FPUA emulsion also exhibited a good mechanical stability and the Zeta potentials were all below −27 mV. The investigation of UV-curing kinetics indicated that the UV-curing speed had a slightly decrease with the increment of fluorine content. However, the contact angles of the annealed films increased greatly by the increment of fluorine content. Moreover, the bulk morphology characterized by SEM shown that the phase separation was affected by the miscibility of the fluorine chains, the soft segment and hard segment, while anneal had an adverse effect on the phase separation.
References
Kim CK, Kim BK (1991) J Appl Polym Sci 43:2295
Chen GN, Chen KN (1999) J Appl Polym Sci 71:903
Lim CH, Choi HS, Noh ST (2002) J Appl Polym Sci 86:3322
Kim HD, Kim TW (1998) J Appl Polym Sci 67:2153–2162
Yang JW, Wang ZM, Zeng ZH, Chen YL (2002) J Appl Polym Sci 84:1818
Lim H, Lee Y, Park IJ, Lee SB (2001) J Colloid Interface Sci 241:269
Li GH, Li XR, Shen YD, Ren QH (2006) J Appl Polym Sci 99:2721
Jeong HY, Lee MH, Kim BK (2006) Colloids Surf A Physicochem Eng Aspects 290:178
Su T, Wang GY, Xu XD, Hu CP (2006) J Polym Sci Part A Polym Chem 44:3365
Zhu MJ, Qing FL, Meng WD (2008) J Appl Polym Sci 109:1911
Li H, Zhang ZB, Hu CP (2004) Eur Polym J 40:2195
Dai JB, Zhang XY, Chao J (2007) J Coat Technol Res 4:283
Tanaka H, Suzuki Y, Yoshino F (1999) Colloids Surf A Physicochem Eng Aspects 153:597
Jiang M, Zhao XL, Ding XB (2005) Eur Polym J 41:1798
Ashish V, Manoj KC (2002) J Colloid Interface Sci 249:235
Xu B, Li L, Yekta A, Masoumi Z, Kanagalingam S, Winnik MA, Zhang KW, Macdonald PM (1997) Langmuir 13:2447
Demien C, Andre C, Michel V (2003) Macromolecules 36:449
Bongiovanni R, Gianni AD, Priola A, Pollicino A (2007) Eur Polym J 43:3787
Bongiovanni R, Malucelli G, Messori M (2000) J Appl Polym Sci 75:651
Wang ZM, Yang JW, Chen YL (1999) J Appl Polym Sci 73:2869
Asif A, Shi WF (2004) Polym Adv Technol 15:669
Bai CY, Zhang XY, Dai JB (2007) Prog Org Coat 59:331
Rekha N, Asha SK (2008) J Appl Polym Sci 109:2781
Lee MH, Jang MK, Kim BK (2007) Eur Polym J 43:4271
Jiang M, Zheng ZH, Ding XB (2007) Colloid Polym Sci 285:1049
Bai CY, Zhang XY, Dai JB (2008) J Polym Res 15:67
Bai CY, Zhang XY, Dai JB, Wang JH (2008) J Coat Technol Res 5:251
Kim YS, Lee YS, Ji Q, McGrath JE (2002) Polymer 43:7161
Tonelli C, Messori M, Toselli M (2001) Polymer 42:9877
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Liu, T., Pan, X., Wu, Y. et al. Synthesis and characterization of UV-curable waterborne polyurethane acrylate possessing perfluorooctanoate side-chains. J Polym Res 19, 9741 (2012). https://doi.org/10.1007/s10965-011-9741-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10965-011-9741-0