
Abstract
A new series of N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g were synthesized by the condensation reaction of bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxylic dianhydride 1 with two equimolars of various amino acids such as L-alanine 2a, L-valine 2b, L-leucine 2c, L-isoleucine 2d, L-phenyl alanine 2e, L-2-aminobutyric acid 2f and L-histidine 2g in an acetic acid solution. Also 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 was synthesized by using a two-step reaction. At first 1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one 6 was prepared from the reaction of two equimolars 4-nitrobenzaldehyde 5 and one equimolar acetone 4 in ethanol and NaHCO3 and dinitro compound 6 was reduced by using Na2S. Then seven new photosensitive and optically active organo-soluble poly(amide–imide)s (PAIs) 8a–g with good inherent viscosities were synthesized from the direct polycondensation reaction of new N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g with 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 by two different methods such as direct polycondensation in a medium consisting of N-methyl-2-pyrrolidone (NMP)/triphenyl phosphite (TPP)/calcium chloride (CaCl2)/pyridine (py) and direct polycondensation in a tosyl chloride (TsCl)/pyridine (py)/N,N-dimethylformamide (DMF) system. The polymerization reactions produced a series of photosensitive and optically active organo-soluble PAIs with high yield and good inherent viscosity. The resulted polymers were fully characterized by means of FTIR and 1H-NMR spectroscopy, elemental analyses, inherent viscosity, specific rotation, solubility tests, UV-vis spectroscopy, differential scanning calorimeter (DSC), thermogravimetric analysis (TGA), and derivative of thermaogravimetric (DTG). These macromolecules exhibited maximum UV-vis absorption at around 370 and 265 nm in a DMF solution.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The development of heat resistant high performance polymers in the past decades has been quite dramatic and has drawn the attention of many polymer scientists all over the world. Aromatic polyimides have earned a reputation as high-performance materials because of their excellent thermal stability, chemical resistance, and outstanding mechanical properties [1]. However, poor handling and intractable characteristics have been major problems because of their high melting points and insolubility. To extend the utility of such high-performance materials, it has been a long-desired goal to synthesize polyamide–imides that can offer a compromise between excellent thermal stability and processability [2–8].
Polymers containing photosensitive moieties such as cinnamate, chalcone, coumarine, dibenzalacetone and their derivatives both in main chain or side chain [9–12] have attracted great interest because of their potential use in various applications, the foremost of these including devices for optical data storage [13], photoresists [14], and photolithographic assemblies [15]. Moreover, the design and synthesis of polymers highly sensitive to light processing continue to be the most attractive targets because various material properties can be improved by the incorporation of appropriate chemical structures into a polymer backbone.
The synthesis and application of chiral polymers is of particular interest from the viewpoint of material science and newly considered topics. Chiral polymers have found successful uses in chromatographic separation of enantiomers, chiral liquid crystals, non-linear optical devices, optical switches and biomedical devices, etc. [16–25]. A direct and effective way for synthesizing chiral polymers is to introduce chiral elements into the polymer backbone or side chains. The combination of poly(amide–imide)s with chiral elements is of synthetic interest and may also lead to chiral recognition membranes. Recently, we have synthesized optically active polymers by different methods [26–28].
In this article, a series of new photosensitive and optically active organo-soluble PAIs 8a–g containing dibenzalacetone moiety were synthesized by the direct polycondensation reactions of seven chiral N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g with 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 by two different methods such as direct polycondensation in a medium consisting of N-methyl-2-pyrrolidone (NMP)/triphenyl phosphite (TPP)/calcium chloride (CaCl2)/pyridine (py) and direct polycondensation in a tosyl chloride (TsCl)/pyridine (py)/N,N-dimethylformamide (DMF) system.
Experimental
Materials
Bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxylic dianhydride 1 (from Aldrich), L-alanine 2a, L-valine 2b, L-leucine 2c, L-isoleucine 2d, L-phenyl alanine 2e and L-2-aminobutyric acid 2f, L-histidine 2g, 4-nitrobenzaldehyde 5, acetone 4 and tosyl chloride (TsCl; from Merck) were used without previous purification. Solvent: N-methyl-2-pyrrolidone (NMP; from Fluka), pyridine (from Acros), triphenyl phosphite (TPP; from Merck) and N,N-dimethylformamide (DMF; from Merck) were used as received. Commercially available calcium chloride (CaCl2; from Merck) was dried under vacuum at 150°C for 6 hrs.
Techniques
1H-NMR and 13C-NMR spectra were recorded on a Bruker 300 MHz instrument (Germany). Fourier transform infrared (FTIR) spectra were recorded on Galaxy series FTIR 5000 spectrophotometer (England). Spectra of solid were performed by using KBr pellets. Vibration transition frequencies were reported in wave number (cm−1). Band intensities were assigned as weak (w), medium (m), shoulder (sh), strong (s) and broad (br). Inherent viscosities were measured by a standard procedure by using a Technico Regd Trad Mark Viscometer. UV-vis absorptions were recorded at 25°C in the 190–790 nm spectral regions with a Perkin-Elmer Lambda 15 spectrophotometer on DMF solution by using cell lengths of 1 cm. Specific Rotations were measured by an A-Kruss polarimeter. Thermal Gravimetric Analysis (TGA and DTG) data for polymers were taken on a Mettler TA4000 System under N2 atmosphere at rate of 10°C/min and differential scanning calorimeter (DSC) was conducted with a DSC Metller 110 (Switzerland) at a heating and heating rate of 10°Cmin−1 in a nitrogen atmosphere. Elemental analyses were performed were performed by Vario EL equipment by Arak University.
Monomer synthesis
Synthesis of N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g
1 g (4.03 mmol) of bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxylic dianhydride 1, 8.06 mmol of L-amino acids 2a–g, 50 mL of acetic acid and a stirring bar were placed into a 250-mL round-bottomed flask. The mixture was stirred at room temperature for overnight and refluxed for 4 hrs. The solvent was removed under reduced pressure, and the residue was dissolved in 100 mL of cold water, then the solution was decanted and 5 mL of concentrated HCl was added. A white precipitate was formed, filtered off, and dried to give compounds N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g.
Synthesis of 1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one 6
0.324 g (5.6 mmol) of acetone 4, 1.691 g (11.2 mmol) of 4-nitrobenzaldehyde 5, 15 mL of ethanol, and a stirring bar were placed into a 50-mL round-bottom flask. Then this mixture was heated in 50°C for 1 h and 5 mL of NaHCO3 (1%) was added slowly at this temperature and refluxed for 3 hrs. Then the reaction mixture was cooled to ambient temperature and 50 mL cooled water was added. A yellow crude product formed and was collected by filtration, washed thoroughly with water, and dried to afford 1.52 g (84%). Mp: 262-264◦C. FTIR (KBr): 3109 (w), 1670 (m), 1649 (m), 1629 (s), 1604 (s), 1531 (s), 1411 (m), 1344 (s), 1190 (s), 1111 (s), 987 (s), 852 (s), 756 (m), 702 (s), 543 (m) cm−1. 1H-NMR (300 MHz, DMSO-d6, TMS): δ; 8.29–8.31 (d, 4H, J = 8.4 Hz), 8.05–8.08 (d, 4H, J = 8.4 Hz), 7.89–7.95 (d, 2H, J = 16.1 Hz), 7.52–7.58 (d, 2H, J = 16.1 Hz) ppm. Elemental analysis: calculated for C17H12N2O5: C, 62.96; H, 3.73; N, 8.64; found: C, 62.88; H, 3.71; N, 8.23.
Synthesis of 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7
To solution 0.8 g (10.2 mmol) of Na2S and 0.35 g (4.16 mmol) of NaHCO3 in 5 mL water, 10 mL methanol was added. The mixture stirred for 30 min in room temperature. The precipitate was filtered, then the filtrate was added to mixture 0.25 g (0.770 mmol) of 1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one 6, and 15 mL methanol, and stirred for 3 hrs under reflux conditions. The mixture was concentrated using rotary evaporator, and the residue poured into water, a red crude product formed and was collected by filtration, washed thoroughly with water, and dried to afford 0.186 g (91%). MP: 295–297°C. FTIR (KBr): 3331 (m), 3211 (m), 3030 (w), 2926 (w), 1624 (m, sh), 1591 (s), 1514 (s), 1438 (m), 1307 (m), 1282 (m), 1172 (s), 1107 (m), 985 (w), 829 (m), 511 (m) cm−1. 1H-NMR (300 MHz, DMSO-d6, TMS): δ; 7.51–7.57 (d, 2H, J = 16 Hz), 7.43–7.45 (d, 4H, J = 7.3 Hz), 6.90–6.95 (d, 2H, J = 16 Hz), 6.57–6.60 (d, 2H, J = 7.3 Hz), 5.84 (s, br, 4H) ppm. 13C-NMR (300 MHz, DMSO-d6): δ; 187.96, 151.95, 143.09, 130.84, 122.57, 120.76, 114.11 ppm. Elemental analysis: calculated for C17H16N2O: C, 77.25; H, 6.10; N, 10.60; found: C, 77.18; H, 6.06; N, 10.60.
Polymer synthesis
Poly(amide–imide)s 8a–g were synthesized by two different methods that as an example the preparation of PAI 8a explains in the following.
-
Method A:
Direct polycondensation in a medium consisting of N-methyl-2-pyrrolidone (NMP)/triphenyl phosphite (TPP)/calcium chloride (CaCl 2 )/pyridine (Py)
0.107 g (0.275 mmol) of N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-alanine 3a, 0.072 g (0.275 mmol) of 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7, 0.1 g (0.9 mmol) of calcium chloride, 0.84 mL, (3.00 mmol) of triphenyl phosphite, 0.1 mL of pyridine and 1.5 mL N-methyl-2-pyrrolidone (NMP) were placed into a 25-mL round-bottomed flask, which was fitted with a stirring bar. The reaction mixture was heated under reflux at 120°C for 7 hrs. Then, the reaction mixture was poured into 50 mL of methanol and the precipitated polymer was collected by filtration and washed thoroughly with hot methanol and dried at 60°C for 12 hrs under vacuum to leave 0.158 g (93%) pale yellow solid polymer 8a.
-
Method B:
Direct Polycondensation in a Tosyl Chloride (TsCl)/Pyridine (Py)/ N,N-Dimethylformamide (DMF) System
A solution of 0.1 mL pyridine, 0.0.078 g (0.411 mmol) of TsCl after 30 min stirring at room temperature was treated with 0.1 mL, (1.36 mmol) of DMF for additional 30 min. The reaction mixture was added dropwise to a solution 0.053 g (0.137 mmol) of diacid 3a in 0.1 mL of pyridine. The mixture was maintained at room temperature for 30 min, and then to this mixture, a solution 0.036 g (0.137 mmol) of 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 in 0.4 mL of Pyridine was added dropwise and the whole solution was stirred at room temperature for 30 min and at 100°C for 2 hrs. As the reaction proceeded, the solution became viscous, then was precipitated in 20 mL of methanol and filtered off, dried under vacuum to leave 0.066 g (79%) brown solid polymer 8a.
Results and discussion
Synthesis of monomers
The asymmetric diacids 3a–g were synthesized by the condensation reaction of bicyclo[2,2,2]oct-7-ene-2,3,5,6-tetracarboxylic dianhydride 1 with two equimolars of L-alanine 2a, L-valine 2b, L-leucine 2c, L-isoleucine 2d, L-phenyl alanine 2e, L-2-aminobutyric acid 2f and L-histidine 2g in an acetic acid solution (Scheme 1) [28]. The yields and some physical properties of these compounds are shown in Table 1.

Preparation of diacids 3a–g
The chemical structure and purity of the optically active diacids 3a–g were proved by using elemental analysis, FTIR, 1H-NMR and 13C-NMR spectroscopic techniques and these data shown in Table 2.
The FTIR spectra of all N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g showed absorption around 2500 and 3400 cm−1, which was assigned to the COOH groups. Peaks appearing at around 1700–1770 cm−1 (acid C=O and symmetric imide stretching), 1390 and 700 cm−1 (imide characteristic ring vibration) confirmed the presence of imide ring and carboxylic groups in these compounds.
As an example, the 1H-NMR spectrum of diacid 3f showed in Fig. 1. The protons H(a) relevant to O-H carboxylic groups appeared at 12.95 ppm. Peak in 4.33–4.38 ppm as a doublet of doublet which were assigned to the H(c) protons, which is a chiral center, peaks between 0.66–0.71 ppm were assigned to aliphatic CH3(g), peak in 1.79–1.93 ppm were assigned to H(f). Protons relevant to olefin bicyclo ring appeared at 6.01–6.10 ppm, H(b). Peaks between 3.20–3.43 ppm were assigned to H(e) (4H) and H(d) (2H), that appeared in D2O exchange 1H-NMR spectrum. Also the 13C-NMR spectrum of diacid 3f showed 8 signals, including C(b) and C(a) in carboxylic acids and carbonyl imide rings, and C(c) in carbon atoms olefin in bicyclo (Fig. 2). These peaks in 1H-NMR and 13C-NMR spectra along with elemental analyses data confirmed the proposed structure of compound 3f.

1H-NMR spectrum of diacid 3f

13C-NMR spectrum of diacid 3f
Also 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 was synthesized by using a two-step reaction. At first 1,5-bis(4-nitrophenyl)penta-1,4-dien-3-one 6 was prepared from the reaction of two equimolars 4-nitrobenzaldehyde 5 and one equimolar acetone 4 in ethanol and a solution of NaHCO3 (1%, w/v) [30]. Then dinitro compound 6 was reduced by using Na2S (Scheme 2) [31].

Synthesis of diamine 7
The chemical structure and purity of compound 6 were proved with elemental analysis, 1H-NMR and FTIR spectroscopy and compound 7 were proved with elemental analysis, FTIR, 1H-NMR, and 13C-NMR spectroscopy.
The measured results in elemental analyses of these compounds were closely corresponded to the calculated ones, demonstrating that the expected compounds were obtained. The FTIR spectrum of compound 7 showed two peaks at 3331 and 3211 cm−1, which were assigned to the NH2 groups.
1H-NMR spectrum of compound 7 showed peaks as a doublet of doublet at 7.43–7.45 ppm and 6.577–6.601 ppm were assigned to the H(b) and H(d) related to aromatic protons and peak as a doublet of doublet at 7.51–7.57 ppm and 6.90–6.95 ppm were assigned to the H(a) and H(c) related to olefin protons. Also a broad singlet peak at 5.84 ppm which was assigned to the H(e) protons of the NH2 groups in this compound (Fig. 3). Also 13C-NMR spectrum of compound 7 showed seven different carbon atoms (Fig. 4).

1H-NMR spectrum of diamine 7

13C-NMR spectrum of diamine 7
Polymer synthesis
The direct polycondensation of a dicarboxylic acid and diamine is one of the well-known methods for PAI synthesis. In this article, we synthesized PAIs 8a–g containing dibenzalacetone moiety by the direct polycondensation reactions of seven chiral N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g with 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 by two different methods such as direct polycondensation in a medium consisting of N-methyl-2-pyrrolidone (NMP)/triphenyl phosphite (TPP)/calcium chloride (CaCl2)/pyridine (py) (method A, Scheme 3) and direct polycondensation in a tosyl chloride (TsCl)/pyridine (py)/N,N-dimethylformamide (DMF) system (method B, Scheme 3).

Synthesis of PAIs 8a–g by two different methods
In method A for direct polycondensation used TPP/Py/CaCl2 as activating agent according to a typical procedure that was shown in Scheme 3. A triphenyl phosphite (TPP)-activated polycondensation (phosphorylation reaction) technique for the synthesis of polyamides was reported by Yamazaki et al [32].
The syntheses and some physical properties of these new PAIs 8a–g are given in Table 3. The entire polycondensation reaction readily proceeds in a homogeneous solution, tough and stringy precipitates formed when the viscous PAIs solution was obtained in good yields. Also the resulting polymers have a range of color between pale yellow and yellow (Table 3).
In method B for the direct polycondensation of diacids 3a–g and aromatic diamine 7, a Vilsmeier adduct was prepared by dissolving TsCl in a mixed solvent of Py and DMF. The polycondensation was carried out as the following way: TsCl was dissolved in Py and after a certain period of time (aging time), the solution was treated with DMF for 30 min. The reaction mixture was added to a solution of diacid in Py. After 30 min, a solution of diamine in Py was added and the whole solution was maintained at room temperature, and then elevated temperature for 2 hrs [16].
Less than this time, the polymers obtained will have lower inherent viscosities, and more than this time, the materials will be degraded. The syntheses and some physical properties of these new PAIs 8a–g are given in Table 4. The entire polycondensation reaction readily proceeds in a homogeneous solution, tough and stringy precipitates formed when the viscous PAIs solution was obtained in moderate yields. Also the resulting polymers have a range of color between red and brown (Table 4). Although PAIs 8a–g obtained in a shorter period by method B, but these polymers obtained with higher inherent viscosities and good yields by method A.
One of the main methods for synthesis of optically active polymers is incorporation of chiral segments in the monomer structure. In this work we used of chiral amino acid moieties for synthesis of chiral diacids 3a–g. Also resulting polymers due to presence chiral amino acid moieties 2a–g in the polymer backbone are optically active and Specific Rotations measured at a concentration of 0.5 g/dL in DMF at 25°C (Tables 3 and 4).
Polymer properties
The elemental analyses of the resulting PAIs 8a–g were in good agreement with the calculated values for the proposed structure (Table 5).
Solubility of the PAIs
One of the main objectives of this study was producing modified poly(amide–imide)s with improved solubility. The incorporation of monomers with dibenzalacetone moiety prevents an efficient molecular packing and restricts the formation of interchain hydrogen bonds, which are responsible of the PAIs intractability. Also these polymers are expected to have higher solubility. The solubility of PAIs 8a–g was investigated as 0.01 g of polymeric sample in 2 mL of solvent. Remarkably, all of these PAIs were easily soluble at room temperature in aprotic polar solvents such as N-methyl-2-pyrrolidinone (NMP), N,N-dimethylacetamide (DMAc), N,N-dimethylformamide (DMF), soluble on heating at 70°C and partially soluble on heating at 70°C in tetrahydrofuran (THF), pyridine (Py) and cyclohexanone, and insoluble in solvents such as acetone, chloroform, ethanol and methanol (Table 6).
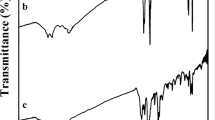
The structure of these polymers was confirmed as PAIs by means of FTIR spectroscopy and elemental analyses. The representative FTIR spectrum of PAI 8d was shown in Fig. 5. The polymer exhibited characteristic absorption bands at 1772 cm−1 for the imide ring (asymmetric C=O stretching vibration), 1709 cm−1 (symmetric C=O stretching and amide stretching vibration) in the main chain, 1379 cm−1 (C-N stretching vibration). The absorption bands of amide groups appeared at 3352 cm−1 (N-H stretching). FTIR characterizations of all PAIs are given in Table 7.

FTIR spectrum of PAI 8d
The 1H-NMR spectra of PAIs 8b and 8f showed peaks that confirm their chemical structure that display in Figs. 6 and 7. Figure 6 displays 1H-NMR spectrum of PAI 8b that the aromatic protons and four olefin protons related diamine 7 appeared in the region of 7.21–7.72 ppm and the peak in the region of 10.02 ppm is assigned for N-H amide groups in the polymer backbone. Also Fig. 7 displays 1H-NMR spectrum of PAI 8f that the aromatic protons and four olefin protons related diamine 7 appeared in the region of 7.22–7.73 ppm and the peak in the region of 9.97 ppm is assigned for N-H amide groups in the polymer backbone.

1H-NMR spectrum of PAI 8b

1H-NMR spectrum of PAI 8f
UV-vis absorption characteristics
The photosensitive property of the new poly(amide–imide)s 8a–g in the DMF solution was studied by a UV-vis spectrophotometer. Due to presence of dibenzalacetone moiety in the polymer backbone, these PAIs have photosensitive properties [11, 33]. All polymer solutions exhibit two same positions of absorption maximum in UV-vis spectra around 370 nm and 265 nm.
The absorption maximum at around 265 nm corresponds to π→π* transition of the olefinic double bond present in the dibenzalacetone moiety and carbon double bonds in aromatic rings in the polymer backbone. Also the absorption maximum at around 370 nm corresponds to n→π* transition of the nonbonding electrons present in nitrogen and oxygen atoms in the polymer backbone. The UV-vis absorption spectrum of PAI 8f in N,N-dimethyl formamide is shown in Fig. 8. The spectrum of PAI 8f exhibited two typical peaks at 265.71 nm (π→π*) and 369.81 nm (n→π*).

UV-vis absorption spectrum of PAI 8f in DMF solution
Thermal properties
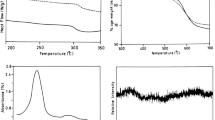
TGA and derivative of thermaogravimetric (DTG) analysis at a rate of 10°Cmin−1 in a nitrogen atmosphere were utilized to examine the thermal properties of these PAIs, and the obtained results are summarized in Table 8. Figure 9 show TGA results of PAIs 8a, 8b and 8e, respectively.

TGA curves of PAIs 8a, 8b and 8e
The thermal stability of the polymers was studied on the basis of 5 and 10% weight losses (T5 and T10, respectively) of the polymers and the residue at 800°C (char yield). The results revealed that the PAIs were thermally stable up to 310°C. TGA data showed that the resulting polymers were good thermally stable. Also the DSC analyses for PAIa showed Tg around 157–173°C (Table 8).
The char yield can be applied as a decisive factor for estimated the limiting oxygen index (LOI) of polymers according to the equation of Van Krevelen and Hoftyzer [29]:
Where CR is the char yield.
PAIs 8a, 8b and 8e had LOI values around 30, which were calculated from their char yield. On the basis of the LOI values, such macromolecules can be classified as self-extinguishing polymers.
Conclusions
A series of new photosensitive and optically active organo-soluble PAIs 8a–g containing dibenzalacetone moiety were synthesized by the direct polycondensation reactions of seven chiral N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids 3a–g with 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one 7 by two different methods. The photosensitive property of the new poly(amide–imide)s 8a–g in the DMF solution was studied by a UV-vis spectrophotometer. Due to presence of dibenzalacetone moiety in polymer backbone, these PAIs are photosensitive. Because the resulting polymers contained optically pure L-amino acid moieties, they showed optical rotations and were optically active. Because of presence of dibenzalacetone moiety in polymer chain, these polymers are expected to have higher solubility. Potential applications of amino acid based polymers include drug delivery agents, chiral stationary phases for the resolution of enantiomers in chromatographic techniques, and biomaterials.
References
Ghosh MK, Mittal KL (1996) Polyimide: fundamentals and applications. Marcel Dekker, New York
Yang CP, Chen RS, Chen SH (2001) J Polym Sci Part A Polym Chem 39:93–104
Yang CP, Su YY (2005) J Polym Res 12:17–22
Liaw DJ, Chen WH (2003) Polymer 44:3865–3870
Liaw DJ, Chang FC, Liu JH, Wang KL, Faghihi Kh, Huang SH, Lee KR, Lai JY (2007) Polym Degrad Stabil 92:323–329
Faghihi Kh, Hajibeygi M (2004) J Appl Polym Sci 92:3447–3453
Faghihi Kh, Hagibeygi M (2005) Macromol Res 13:14–18
Yang CP, Chen YP, Woo EM (2004) Polymer 45:5279–5293
Mihara T, Nakao Y, Koide N (2005) Polym J 36:899–908
Lee SW, Ree M (2003) J Polym Sci Part A Polym Chem 42:1322–1334
Balakrishnan PS, Murugavel SC (2009) J Appl Polym Sci 111:2340–2344
Feng K, Tsushima M, Matsumoto T, Kurosaki T (1998) J Polym Sci Part A Polym Chem 36:685–693
Natansohn A, Rochon P (2002) Chem Rev 102:4139–4175
Gupta P, Trenor SR, Long TE, Wilkes GL (2004) Macromolecules 37:9211–9218
Morigaki K, Schonherr H, Frank CW, Knoll W (2003) Langmuir 19:6994–7002
Mallakpour S, Kolahdoozan M (2007) J Appl Polym Sci 104:1248–1254
Okamoto YE, Yashima E (1998) Angew Chem Int Ed Engl 37:1020–1043
Chiellini E, Galli G, Dossi E, Cioni F (1993) Macromolecules 26:849–851
Fujishiro K, Lenz RW (1992) Macromolecules 25:81–87
Ciardelli F (1987) Encyclopedia of polymer science and engineering. Wiley, New York
Jager WF, Jong JC, Lange B, Huck NPM, Meetsma A, Feringa BL (1995) Angew Chem Int Ed Engl 34:348–350
Feringa BL, Jager WF, Lange B (1991) J Am Chem Soc 113:5468–5470
Thiem J, Bachmann F (1993) Makromol Chem 194:1035–1057
Lenz RV, Guerin P (1983) Polym Sci Technol 23:219–224
Mallakpour SE, Hajipour AR, Faghihi K (2001) Eur Polym J 37:119–124
Faghihi Kh, Zamani Kh, Mirsamie A, Mallakpour S (2004) J Appl Polym Sci 91:516–524
Faghihi Kh (2004) Macromol Res 12:258–262
Faghihi Kh (2008) J Appl Polym Sci 109:74–81
Van Krevelen DW, Hoftyzer PJ (1976) Properties of Polymers. Elsevier, New York
Zhang Z, Dong YW, Wang GW (2003) Chem Lett 32:966–967
Shriner RL, Hermann CKF, Morrill TC, Curtin DY, Fuson RC (1997) The systematic identification of organic compounds. Wiley, New York
Yamazaki N, Matsumoto M, Higashi F (1975) J Polym Sci Polym Chem 13:1373–1380
Guo M, Wang X (2009) Eur Poly J 45:888–898
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Faghihi, K., Hajibeygi, M. & Shabanian, M. New photosensitive and optically active organo-soluble poly(amide–imide)s from N,N′-(bicyclo[2,2,2]oct-7-ene-tetracarboxylic)-bis-L-amino acids and 1,5-bis(4-aminophenyl)penta-1,4-dien-3-one: synthesis and characterization. J Polym Res 17, 379–390 (2010). https://doi.org/10.1007/s10965-009-9324-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10965-009-9324-5