
Abstract
Nylon 11/ethylene-vinyl alcohol (EVOH)/dicumyl peroxide (DCP) blends were prepared using a single-screw extruder. The Fourier transcript infrared spectroscopy results showed that strong interactions existed and co-crystallization formed between nylon 11 and EVOH in the presence of DCP. The crystalline morphology and melting behavior was investigated by means of Wide-angle X-ray diffraction (WAXD), polarized optical microscope (POM) and differential scanning calorimetry, respectively. The WAXD indicated that no crystal structure transition occurred for both nylon 11 and EVOH after the addition of DCP. The spherulites size of nylon 11 became smaller and crystalline bands of EVOH formed in the presence of DCP as revealed by POM observation. Both nylon 11 and its blends exhibited two melting endotherms. The equilibrium melting point for blends was lower than that of nylon 11, it was indicated that the mobility of nylon 11 was hindered after the addition of EVOH and DCP.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Since the permeation of hydrocarbon solvents through plastic fuel tanks and agrochemical bottles created pollution, safety, and health problems, more attentions had been paid on the manufacture of high-barrier plastic containers. In recent years, alternative technologies, including surface treatment by fluorination or sulfonation, multilayer coextrusion and laminar-blend blow molding, have been applied to overcome the permeation problems. Since laminar-blend-blow molding process lends itself to a wide variety of container designs and to processing convenience, there has been considerable interest in studying laminar barrier technology in the last decades [1–4]. In many published literatures [5–7], nylon 11/ethylene-vinyl alcohol (EVOH) and nylon generally has been chosen as the barrier layer for the low barrier performance materials. It is well recognized that nylon has good barrier properties against hydrocarbon solvent and exhibits outstanding levels of toughness while, at the same time, it offers good adhesion to a variety of resinous substrates or coating. In comparison with nylon, EVOH exhibits even better hydrocarbon permeation resistance and is recognized as an oxygen-barrier material in food and other packaging applications that require protection from oxygen, due to its superior gas barrier property and high oil resistance. However, EVOH were affected by its brittleness and moisture-absorbance. Moreover, EVOH generally have a poor compatibility with other polymers whether of polar or non-polar nature [8]. With the aims of boosting the barrier property of nylon or improving the mechanical properties and/or adhesion of EVOH, melt-mixed blends of nylon and EVOH have been investigated prodigiously over the past few years [9–11]. Although EVOH has been shown some miscibility with nylon in some literatures [12–15], blending of EVOH into nylon had been demonstrated to deteriorate the mechanical properties of nylon matrix [16, 17].
It is well known that physical, mechanical and barrier properties of crystalline polymer depend on the morphology, crystalline structure and degree of crystallinity. We developed a new in-situ co-crosslinking technology to control the EVOH dispersion phase morphology and to improve the interfacial adhesion between EVOH and nylon 11, both barrier property and mechanical properties has been proven to be enhanced. In our previous paper [18], we discussed the cocrosslinking mechanism between nylon 11 and EVOH in the presence of dicumyl peroxide (DCP). The peroxide decomposed to RO· radicals, and these radicals abstracted hydrogen from both EVOH and nylon 11 to yield free radicals. The free radical is next to the carbonyl group in nylon 11, so β-scission of nylon 11 radical occurred to yield a primary radical, which readily accessed the tertiary radicals of EVOH, and thus formed cocrosslinking. In this paper, the crystalline morphology and melting behavior of nylon 11/EVOH/DCP blends were investigated by Wide-angle X-ray diffraction (WAXD), polarized optical microscope (POM) and differential scanning calorimetry (DSC). The interactions between nylon 11 and EVOH was also characterized by Fourier transform infrared spectroscopy (FTIR).
Experiment
Materials and preparation
Nylon 11 was prepared by our laboratory, with weight-average \({\left( {\overline{M} _{w} } \right)}\) and number-average \({\left( {\overline{M} _{n} } \right)}\) molecular Weights of 19.8 × 104 and 4.0 × 104 gmol−1, respectively. The EVOH (F101B) was obtained from Kuraray Co, Ltd (Japan), ethylene content, 32 mol%. DCP was supplied by Gaoqiao Petrochemical Corporation (Shanghai, China). Nylon 11 and EVOH was dried under vacuum at 80 °C for 12 h and kept in desiccator prior to use. Melt blends were obtained by using a SJ20/25 single-screw extruder (China). The different components were fed together under the screw speed of 70 rpm at temperatures 190, 220 and 220 °C from hopper to die, respectively. The extrudate passed through a cooling water bath and were finally palletized, and then dried in vacuum at 80 °C for 24 h. In this paper, the neat nylon 11 and EVOH are abbreviated as itself; and the nylon 11/EVOH (nylon 11/EVOH = 80/20) blends containing 0% DCP and 1.5%DCP are abbreviated as N-20E-0D and N-20E-1.5D, respectively.
Fourier transform infrared spectroscopy
A SHIMADZU 8400S spectrometer was used to record spectra using a spectral of 2 cm−1. Thin film samples were prepared using with dimensions molding in a press at a temperature of 230 °C, rapidly cooled to 166 °C, stay there for 1 h until crystallization was complete, and then cooled to room temperature by air.
Wide-angle X-ray diffraction
The WAXD measurement was obtained in a Rigaku D/Max-Ra diffractometer with curved graphite crystal filtered. The corresponding wide-angle X-ray diffraction patterns were collected in transmission mode at room temperature operating under a tube current of 100 mA and an acceleration voltage of 40 kV. The scanning rate was 4°/min. The samples for measurement were prepared as the same as that for FT-IR.
Polarized optical microscope
The crystallite morphologies were observed by XPT-7 POM (China). In order to observe and compare with the crystal morphology of samples clearly during the isothermal crystallization process, we choose the middle temperature of the range of isothermal crystallization experiment to observe by POM. The samples, placed on glass slides covered with slips, isothermally crystallized for 2 h at the temperature of 164 °C, and then cooled to room temperature by air. All the samples were observed at magnification of ×630.
Differential scanning calorimetry
A METTLER DSC822e, flushed with nitrogen, was used to record crystallization exotherms and melting endotherms. The temperature was calibrated with the indium standard. Sample weights were between 2 and 3 mg. All samples were dried at 80 °C under vacuum for 24 h.
DSC experiment of isothermal crystallization and subsequent melting behaviors were performed as follow: the sample were heated to 230 °C at a rate of 50 °C/min and held at this temperature for 10 min to eliminate any previous thermal history, and then cooled at a rate of −100 °C/min to predetermined crystallization temperature (T c ), raged from 160 to 170 °C in steps of 2 °C and was maintained at T c for 1 h necessary for the DSC trace to return to calorimeter baseline. The specimens were subsequent heated to 230 °C at a rate of 10 °C/min.
The samples for non-isothermal crystalline were rapidly heated at a rate of 100 °C/min to 230 °C, stayed there for 10 min to eliminate any residual crystals, and then cooled to −30 °C at different cooling rate of 2.5,5,10,20 and 40 °C/min, then heated to 230 °C at a rate of 10 °C/min.
Results and discussion
Interactions between components revealed by FTIR
The curves a–d in Fig. 1 presented the FT-IR spectra of nylon 11, nylon 11/EVOH, nylon 11/ EVOH/DCP blends, and EVOH, respectively. As a reference, the FT-IR spectra of the blends were compared with that of pure nylon 11 and EVOH. The spectra of the blends showed a new peak appeared at 1,736 cm−1, which was assigned to carbonyl (C=O) in ester group. It was indicated that chemical reaction existed between hydroxyl groups in EVOH and terminal carboxyl groups in nylon 11. This result was in agreement with the report by Artzi [9]. Additionally, the position of typical polyamide peaks (3301 cm−1 for N—H stretching, 2916 cm−1 for C—H stretching, 1627 cm−1 for C=O stretching, 1542 cm−1 for C—N bending) was found to be shifted to some extent, which indicated that hydrogen bonding existed in the blends. It was in agreement to the report by Chang-Sik Ha [17]. In comparison with the spectra of nylon 11/EVOH blends, there was no new typical peak appeared for the nylon 11/EVOH/DCP blends, but the position of above-mentioned typical peaks shifted to higher wavenumbers. It was contributed to the β-scission of nylon 11 initiated by the addition of DCP. This result further confirmed the cocrosslinking mechanism reported in our previous work [18].

FT-IR spectra of nylon 11, nylon 11/EVOH, nylon 11/ EVOH/DCP blends, and EVOH
Figure 2 illustrated the FT-IR trace between 600 and 800 cm−1 of Fig. 1 in more detail. As can be seen from Fig. 2a, there were double peaks at 729.45 and 719.40 cm−1 which were attributed to rocking model of CH 2 methylene units in nylon 11 chain. However, as shown in Fig. 2b and c, only single peak was observed at 721.33 and 721.61 cm−1, respectively. It was speculated that transition from double peaks to single peak is the character of co-crystallization. The occurrence of co-crystallization can be interpreted as follows: (1) both nylon 11 and EVOH have common chain segment –(CH2) n –, which can tangle with each other under exogenic force, and (2) the double peaks arises from the interactions between pairs of like groups in adjacent like stems; the single component arises either from a CH2 stem isolated from like stems, surround by stems of the other type or from like stems arrayed in a regular manner along the (200) plane, which was shown by WAXD in Fig. 4. The splitting width of the double peaks depends on the content of co-crystallization. As the content of co-crystallization increased, the splitting double peaks turned into single peak gradually [19, 20].

FT-IR trace between 600 and 800 cm−1 of Fig. 1 in more detail
WAXD analysis
Figure 3 showed the WAXD patterns for the pure nylon 11, N-20E-0D, N-20E-1.5D and the pure EVOH. It can be seen that the nylon 11 exhibited two strong reflections (100) and (010/110) at 2θ of 20.14° and 23.10°, which were characteristic of the triclinic α-form. On the other hand, the pure EVOH specimen exhibited an monoclinic α-form crystals diffraction pattern with four peak diffraction angles at 10.8°, 19.78°, 20.4°, 22.3° [1, 11], respectively. Generally, the similar crystal structure could reduce the free energy of co-crystallization, it was thus favorable for the occurrence of co-crystallization between nylon 11 and EVOH. The characteristic diffraction patterns of monoclinic α-form EVOH crystals could also be found in nylon11/EVOH blends whereas became less demarcative after blending with nylon 11. In the case of the addition of DCP, it was found that both triclinic α-form in nylon 11 and monoclinic α-form in EVOH became weaken. It might be owing to that the crystallization was impeded after the addition of DCP.

WAXD patterns for the pure nylon 11, N-20E-0D, N-20E-1.5D and the pure EVOH
Crystal morphology
The POM was used to characterize the crystallization morphology of polymer and its blends. Figure 4a–c presented the POM micrographs pure nylon 11, N-20E-0D and N-20E-1.5D, respectively. As can be seen from Fig. 4a, the nylon 11 displayed the usual spherulite structure typified by Maltese cross. However, a dense granular texture of crystals was formed in the presence of EVOH and the size of spherulite became smaller (illustrated as Fig. 4b). The EVOH permeated through nylon 11 matrix and tangled with nylon 11 macromolecular chains, and thus restricted the ordered rearrangement of the nylon 11 chains. On the other hand, the combination of the strong chemical reaction and the hydrogen bonding, which was confirmed by FTIR, also impeded the mobility of the nylon 11 chains. Therefore, it resulted in the formation of small and imperfect spherulites. After the addition of DCP, as shown in Fig. 4c, the blends was found to be partially cross-linked and typical spherulites of nylon 11 could hardly be observed. However, it was worthy noticeable that some laminar crystalline domains appeared. As reported in our previous work [18], the crosslinking reaction resulted in the increasing viscosity of the EVOH and improved the interfacial adhesion between EVOH and nylon 11. The opportune DCP concentration made the melt strength of the EVOH not too hard or too soft, so it favored the formation of well-developed laminar morphology, and EVOH was dispersed within nylon 11 in the form of discontinuous mutual crossover laminar structure. Take the fact that EVOH was a readily crystalline polymer into consideration, we argued that these laminar crystalline domains were aggregates of numerous microcrystallites of EVOH.

a–c POM micrographs pure nylon 11, N-20E-0D and N-20E-1.5D
Melting behavior of crystallization
Melting behavior of isothermal crystallization
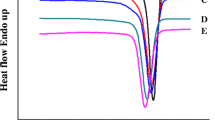
Figure 5a–c presented a series of DSC heating thermograms for the pure nylon 11, N-20E-0D and N-20E-1.5D, respectively. All specimens were melt-crystallized at different T c . As can be seen, the DSC endotherms for all the specimens exhibited two melting peaks. The melting peak at low temperature shifts towards to the higher temperature while the melting peak at high temperature displays almost the same with increasing crystallization temperature, and moreover, the two peaks merge into a single one in the case of T c higher than 168 °C. Such behavior can be attributed to the re-crystallization or different thermal history [21–24]. The melting peak at low temperature is assumed to be due to the melting of crystals formed on cooling from melt, while the second peak, at low T c ′s, is mainly due to melting of crystals which have re-crystallized during the heating run (after the first melting peak). Similar results have been reported for the blends of various functionalized polyolefins and nylon 6 by Wei et al. [24].

a–c Series of DSC heating thermograms for the pure nylon 11, N-20E-0D and N-20E-1.5D
Equilibrium melting point
To perform the quantitative analysis of crystallization behavior, especially to investigate the temperature dependence of the crystalline rate, it is necessary to determine the equilibrium melting point. According to Hoffman-weeks theory, the equilibrium melting point \({\left( {T^{0}_{m} } \right)}\) can be deduced by plotting the melting temperature (T m ) versus the crystallization temperature (T c ). The \({\left( {T^{0}_{m} } \right)}\) was obtained by extrapolation of the resulted straight line to intersect the line T m = T c . Mathematically, the dependence of T m on T c was expressed as
Where β was the lamellar thickening factor which described the growth of lamellar thickness during crystallization and was supposed to be greater than or equal to 1. The lower temperature and higher temperature as a function of T c for all specimens were plotted in Fig. 6. The linear extrapolation of the experimental data up to T m = T c line was also drawn. The extrapolation of T m versus T c showed that the melting temperature data corresponding to the lower endotherms were well aligned with those obtained when only one melting peak was observed. However, the high-temperature peaks were at approximately the same position. In spite of the changed crystalline temperature, the lower endotherm temperature was used to calculate the \(T^{0}_{m} \). The \(T^{0}_{m} \) value of 200.95, 198.22 and 197.00 °C for the pure nylon11, N-20E-0D and N-20E-1.5D was obtained, respectively. The \(T^{0}_{m} \) value of the pure nylon 11 was in agreement with that reported by Liu SY [25] and Zhang XK [26]. Additionally, the \(T^{0}_{m} \) values for blends were lower than that for pure nylon 11, indicating that the crystals in blends were less perfect than those in pure nylon 11.

The lower temperature and higher temperature as a function of T c for all specimens
Melting behavior of non-isothermal crystallization
All the specimens of nylon 11 and its blends were cooled from the melt at different rate of −2.5, −5, −10, −20 and −40 °C/min, and then the melting traces were recorded. The melting traces for pure nylon 11, N-20E-0D and N-20E-1.5D were shown in Fig. 7a–c, respectively. As can be observed, similar with the melting behavior for isothermal crystallization, there were also two melting peaks for non-isothermal crystallization. Both peaks shifted toward lower temperatures as the cooling rates increased. It was due to that there was no enough time for the molecular chain rearrangement at high cooling rate, and then more imperfect crystals were formed. It was worthy noted that an additional melting peak at about 203 °C appeared in Fig. 7c, which were also observed in Fig. 4c. We speculated that the new peak at about 203 °C was contributed to the melt of co-crosslinked macromolecules between nylon 11 and EVOH initiated by addition of DCP.

a–c Melting traces for pure nylon 11, N-20E-0D and N-20E-1.5D
Conclusion
The crystalline morphology and melting behavior of pure nylon 11 and its blends were investigated by FTIR, WAXD, POM and DSC. Some strong interactions (such as chemical reaction and hydrogen bonding) existed between nylon 11 and EVOH, and co-crystallization occurred in the presence of DCP as revealed by transition from double peaks to single peak at about 721 cm−1. No crystalline structure transition was found for both nylon 11 and EVOH after the addition of DCP, however the spherulites size of nylon 11 was decreased and crystalline band of EVOH formed in the presence of DCP. There were two melting endotherms for both isothermal and non-isothermal crystallization process. Especially, a new melting peak, which assigned to the melting of co-crosslinked macromolecules between nylon 11 and EVOH, was found at about 203 °C. The equilibrium melting point for blends was lower than that for nylon 11, it was indicated that the addition of EVOH and DCP resulted in the formation of imperfect nylon 11 spherulites, which also were confirmed by POM observation.
Reference
Yeh J-T, Yao W-HQ et al (2005) J Polym Sci 43:511
Yeh J-T, Huang S-S, Chen H-Y (2005) J Appl Polym Sci 97:1333 doi:10.1002/app.21837
Lagaron JM, Gimenez E, Catala R et al (2005) Macromol Chem Phys 204:704 doi:10.1002/macp.200390039
Montoya M, Abad MJ, Barral L et al (2006) Eur Polym J 42:265 doi:10.1016/j.eurpolymj.2005.08.003
Huang C-H, Wu J-S, Huang C-C et al (2004) J Polym Res 11:75 doi:10.1023/B:JPOL.0000021796.41800.e9
Russo P, Acierno D, Di Maio L et al (1999) Eur Polym J 35:1261 doi:10.1016/S0014-3057(98)00197-9
Faisant JB, Aït-Kadi A, Bousmina M et al (1998) Polymer (Guildf) 39:533 doi:10.1016/S0032-3861(97)00313-3
Coleman MM, Yang X, Zhang H et al (1993) J Macromol Sci- Phys B32:295
Artiz N, Khatua BB, Tchoudakov R et al (2004) J Macromol Sci B43:605
Lagaron JM, Gimenez E, Gavara R et al (2001) Polymer (Guildf) 42:9531 doi:10.1016/S0032-3861(01)00496-7
Lagaron JM, Gimenez E, Saura JJ et al (2001) Polymer (Guildf) 42:7381 doi:10.1016/S0032-3861(01)00204-X
Lee SY, Kim SC (1998) J Appl Polym Sci 67:2001 doi:10.1002/(SICI)1097–4628(19980321)67:12<2001::AID-APP6>3.0.CO;2-Q
Uno M, Norton LJ, Kramer EJ et al (1998) J Mater Sci 33:853 doi:10.1023/A:1004382905447
Nir Y, Narkis M, Siegmann A (1998) Polym Eng Sci 38:1890 doi:10.1002/pen.10359
Liu Y, Donovan JA (1995) Polymer (Guildf) 36:4797 doi:10.1016/0032-3861(95)99295-6
De Petris S, Laurienzo P, Malinconico M et al (1998) J Appl Polym Sci 68:637 doi:10.1002/(SICI)1097-4628(19980425)68:4<637::AID-APP15>3.0.CO;2-O
Ha C-S, Ko M-G, Cho W-J (1997) Polymer (Guildf) 38:1243 doi:10.1016/S0032-3861(96)00821-X
Ding Z, Hu G, Wang B (2007) J Polym Res 14:511–517 doi:10.1007/s10965-007-9136-4
Morgan RL, Hill MJ, Barham PJ (1999) Polymer (Guildf) 40:337 doi:10.1016/S0032-3861(98)00193-1
Tashiro K, Satkowskim M, Steinrs RS et al (1995) Macromolecules 28(8):477
Li YJ, Zhu XY, Tian GH, Yan DY, Zhou EL (2001) Polym Int 50:677 doi:10.1002/pi.682
Lee Y, Porter RS (1989) Macromolecules 22:1754
Li J, Organ SJ, Hobbs JK, Terry AE, Barham PJ, Seebach D (2004) Polymer (Guildf) 45:8913 doi:10.1016/j.polymer.2004.10.034
Wei Q, Chionna D, Galoppini E, Pracella M (2003) Macromol Chem Phys 204:1123 doi:10.1002/macp.200390081
Liu S, Cui Y, Zhang H et al (2006) J Appl Polym Sci 70(1998):2371
Zhang X, Xie T, Yang G (2006) Polymer 47:2116 doi:10.1016/j.polymer.2006.01.077
Acknowledgement
This work was financially supported by the Science and Technology Foundation for Younth from Shanxi Sci and Tech Bureau (Grant No. 2006021011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hu, Gs., Ding, Zy., Li, Yc. et al. Crystalline morphology and melting behavior of nylon11/ethylene-vinyl alcohol/dicumyl peroxide blends. J Polym Res 16, 263–269 (2009). https://doi.org/10.1007/s10965-008-9225-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10965-008-9225-z