
Abstract
In this work, thermophysical properties of n-ethylpyridinium bis(trifluoromethylsulfonyl)imide have been studied at atmospheric pressure in the temperature range 288.15–338.15 K. Density, speed of sound, refractive index, surface tension, isobaric molar heat capacity, electrical conductivity and kinematic viscosity have been measured; from these data the isobaric expansibility, isentropic compressibility, molar refraction, entropy and enthalpy of surface formation per unit of surface area, and dynamic viscosity have been calculated. Moreover, we have characterized the thermal behavior of the compound. Results have been analyzed paying special attention to the structural and energetic factors. The magnitude and directionality of the cation–anion interactions have been studied using ab initio quantum calculations, which allow a better understanding of the physicochemical behavior of the ionic liquid. Finally, density values and radial distribution functions were also estimated ab initio from classical molecular dynamics simulations, providing acceptable density predictions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Ionic liquids are formed of large asymmetric cations as pyridinium, imidazolium or phosphonium, together with different organic or inorganic anions, such as tetrafluoroborate, bis(trifluoromethylsulfonyl)imide, dicyanamide or halides. It is possible to combine different cations and anions to design ionic liquids with desirable properties. In this sense, the determination of physicochemical properties of different kinds of ionic liquids can provide information about the ionic structures and intermolecular interactions. This knowledge is necessary for the implementation of these new solvents in industry and novel technologies [1–7].
With this aim, we present here the thermophysical properties of the ionic liquid, 1-ethylpyridinium bis(trifluoromethylsulfonyl)imide ([epy][Tf2N]). We have also found some physicochemical information about this ionic liquid [8–12].
Density, speed of sound, refractive index, surface tension, isobaric molar heat capacity, electrical conductivity and kinematic viscosity have been measured at atmospheric pressure in the temperature range 288.15–338.15 K with a temperature step of 2.5 K. Moreover, the thermal behavior around ambient temperature has been registered. From the experimental results, we have obtained isobaric expansibility, isentropic compressibility, molar refraction, entropy and enthalpy of surface formation per unit of surface area and the dynamic viscosity. In order to establish the influence of the methyl group in the pyridine ring, these results have been compared to those obtained for 1-ethyl-2-methylpyridinium bis(trifluoromethylsulfonyl)imide ([e2mpy][Tf2N]), an ionic liquid with the same anion but different cation, which has been studied previously [13].
In addition, to increase the knowledge about this solvent, we have performed a theoretical study using quantum calculations and classical molecular dynamics simulations. These methods provide a computational alternative to more traditional predictive models of thermodynamic properties.
2 Experimental
The ionic liquid, 1-ethylpyridinium bis(trifluoromethylsulfonyl)imide (purity 99 %) was obtained from IoLiTec. To reduce the water content as much as possible, the IL was dried under a vacuum of about 0.05 kPa during 24 h under stirring and stored before use in a desiccator. The water content of sample was less than 250 ppm as determined by Karl-Fischer titration. On the other hand, the halide content was checked by 19F NMR, the content being less than 100 ppm, which agrees with supplier’s indication.
Densities, ρ, and speed of sounds, u, of the pure compound were determined simultaneously with an Anton Paar DSA 5000 vibrating tube densimeter and sound analyzer, automatically thermostated within ±0.005 K. By measuring the damping of the oscillation of the U-tube caused by the viscosity, the apparatus automatically corrects for viscosity related errors in the density. The calibration was carried out with ultrapure water, supplied by SH Calibration service GmbH, and dry air. The final uncertainty of densities and speeds of sound can be estimated in ±5 × 10−5 g·cm−3 and ±0.05 m·s−1, respectively.
The refractive index, n D, corresponding to the 589.3 nm sodium D wavelength, was measured using a high precision automatic refractometer Abbemat-HP DR Kernchen. The temperature of the sample was controlled within ±0.01 K by a built-in Peltier device, while another Peltier thermostat was used to keep the internal temperature of the refractometer components constant. The apparatus was calibrated with deionized double-distilled water. The corresponding uncertainty is ±5 × 10−6.
The surface tension, σ, of the pure liquid was determined using a drop volume tensiometer Lauda TVT-2. The temperature was kept constant within ±0.01 K by means of an external Lauda E-200 thermostat. The uncertainty of the measurement is ±0.05 mN·m−1 for the final values of surface tension.
The isobaric molar heat capacity, C p , of the IL was determined using a differential scanning calorimeter Q2000 from TA Instruments equipped with a refrigerated cooling system. The melting transition (T = 429.76 K, ΔH = 3.296 kJ·mol−1) of a standard indium sample was used in order to calibrate the temperature and energy. The zero-heat-flow procedure described by TA Instruments has been followed for the heat capacity measurements using a synthetic sapphire sample as a reference compound. All of these experiments allow estimating an overall uncertainty ±0.5 K in temperature and ±3 % in the heat capacities. The measurement was carried out using around 15 mg of sample sealed at room temperature in hermetic aluminum pans with a mechanical crimp. Previously, the sample was submitted to a vacuum drying process and it was exposed to the air for only a few minutes during the sealing step. Once inside the calorimeter, the sample was cooled to T = 278 K and, after thermal equilibration, a heating thermogram was performed at a scan rate of 10 K·min−1 up to T = 343 K.
Values of electrical conductivity, κ, were determined using a conductimeter from CRISON, model GLP31. Measurements were carried out in a sealed and evacuated cell to avoid exposure of the sample to air. The results are averages of three measurements to ensure the reproducibility within 1 % of the absolute value. The temperature of the sample was kept constant within ±0.01 K by means of a Lauda E-200 thermostat and it was measured with a thermometer F250 from Automatic Systems Laboratories. The cell was calibrated with aqueous solutions at different concentrations as supplied by CRISON.
The kinematic viscosity, ν was determined using a set of Ubbelohde viscometers with a Schoot–Geräte automatic measuring unit model AVS-440. The temperature was kept constant within ±0.01 K by means of a Schoot–Geräte thermostat. The viscosimeter constants provided by the supplier are k = 0.3213 and 0.02904 mm2·s−2 and kinetic energy corrections were applied to the measured times. CaCl2 drying tubes were used to protect the sample from moisture in the air. The uncertainty in the kinematic viscosity is ±1 %. From the density and kinematic viscosity the dynamic viscosity was calculated, where the corresponding uncertainty is ±1 %.
DSC experiments were performed in order to study the thermal behavior of the ionic liquid, over the 255–320 K temperature range, using a differential scanning calorimeter Q1000 from TA Instruments. The atmosphere during this process was helium gas. Temperature and enthalpy were calibrated with a standard sample of indium. The ionic liquid was cooled down to 255 K at 10 K·min−1 and then heating up to 320 K at the same scan rate.
3 Results and Discussion
A comprehensive set of properties is an essential tool to find the relationship between structural characteristics of a novel compound and their features. Each property provides important information that leads to an understanding of the liquid state that is very useful from practical and technological points of view. With the aim of finding a relationship between the characteristics of the ionic liquids and ionic structures, we compare the experimental values for each property measured for [epy][Tf2N] with those obtained previously for [e2mpy][Tf2N]. The differences between these liquids were analyzed to determine the effect of the lack of the methyl group on the pyridine ring in the case of [epy][Tf2N].
Values of density, speed of sound, isentropic compressibility, refractive index, molar refraction, surface tension, isobaric molar heat capacity, electrical conductivity and both kinematic and dynamic viscosity of the 1-ethylpyridinium bis(trifluoromethylsulfonyl)imide from T = 288.15 to 338.15 K are gathered in Table S1 of the Supporting Information.
It should be noted that we have measured the liquid properties in the range of temperatures from 288.15 to 338.15 K, despite the fact that there is a solid/liquid transition around the temperatures 293–303 K. Thus, values below 305.65 K are for the supercooled liquid since it can remain as a liquid for long periods of time at temperatures well below its melting point.
Thermophysical properties (density, speed of sound, refractive index, surface tension and isobaric molar heat capacities) have been fitted by means of the following equation:
where Y is the studied property, A and B are the adjustable parameters, and T is the absolute temperature. The fitting parameters are gathered in Table 1.
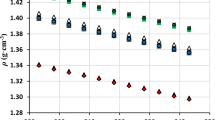
In Fig. 1, the density of the compound is graphically represented. As expected, the density decreases linearly with increasing temperature. Values for [epy][Tf2N] are greater than those found for [e2mpy][Tf2N]. This trend is in agreement with other previous studies [14, 15]. The absence of a methyl group on the pyridine ring leads to higher densities: [epy][Tf2N] has a smaller size and the packing is more efficient than that of [e2mpy][Tf2N] due to the decrease of steric hindrance between ions. The isobaric expansibility, α p = 1/V·(∂V/∂T) p , was calculated from experimental density; this property has a value of 6.44 × 10−4 K−1 at T = 308.15 K which is very similar to the α p value of [e2mpy][Tf2N]. It was reported that most ionic liquids have values between (4 and 7) × 10−4 K−1 [16], suggesting that the densities of ionic liquids have a similar variations with temperature.

The dependence of speed of sound on temperature is shown in Fig. 1. This property decreases when the temperature rises. In this case the values for [epy][Tf2N] are lower than for [e2mpy][Tf2N]. Assuming the ultrasonic absorption is negligible, the isentropic compressibility has been calculated from density and speed of sound using the Newton–Laplace equation, κ S = 1/ρu 2. This increases with temperature and the κ S value at T = 308.15 K is 422.23 TPa−1. The lack of a methyl group makes [epy][Tf2N] more compressible than [e2mpy][Tf2N], therefore it makes structural organization inside the fluid easier.
A plot of the refractive index as a function of temperature is presented in Fig. 2. The measured values decrease as the temperature increases. The experimental values are somewhat smaller for [epy][Tf2N] than for [e2mpy][Tf2N]. Lower values would be expected for [e2mpy][Tf2N] because the packing of the ions is less efficient than in [epy][Tf2N]; however, the refractive index also depends on the medium’s polarizability. When a methyl group is introduced into the pyridinium ring the cation’s polarizability is enhanced leading to an increase of the refractive index of [e2mpy][Tf2N] above the refractive index of [epy][Tf2N].

The molar refraction, R m, has been calculated from the experimental density and refractive index data using the Lorentz–Lorenz relation, R m = V m·[(n 2D − 1)/(n 2D + 2)]. The results show a slight increase of this property with temperature. The molar refraction reaches a value of 66.812 cm3·mol−1 at T = 308.15 K which is smaller than the corresponding value for [e2mpy][Tf2N]. This property represents the hardcore volume of one mole of molecules, and the presence of the methyl group in [e2mpy][Tf2N] makes that the volume occupied by the ions larger.
The surface tension behavior against the temperature is plotted in Fig. 2. As can be seen, this decreases with an increasing temperature. It was found that the σ values of [epy][Tf2N] are slightly smaller than those of [e2mpy][Tf2N] over the whole temperature range. From the experimental surface tensions, we calculated both entropy and enthalpy of surface formation per unit surface area using the following equations, ∆S = (∂σ/∂T) p and ∆H = σ − T(∂σ/∂T) p . The entropy of surface formation per unit area is temperature independent in the investigated temperature range and it has a value of ∆S = 0.05 mN·m−1·K−1. It has been demonstrated using different techniques that [Tf2N]-based ionic liquids show a high degree of surface orientation and consequently they have smaller values of the entropy of surface formation [17]. The ∆S values obtained here are in excellent agreement with those calculated from surface tension measurements of Liu et al. [9], but our values are higher than those of Bittner et al. [11] On the other hand, the enthalpy of surface formation per unit area changes slightly with the temperature and for [epy][Tf2N] has a value of ∆H = 50.75 mN·m−1 at T = 308.15 K while for [e2mpy][Tf2N] the corresponding value is 55.57 mN·m−1; these values are indicative of stronger intermolecular interaction among the fluid constituents in the case of [e2mpy][Tf2N].
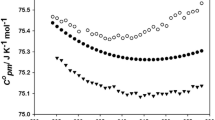
Another useful property, isobaric molar heat capacity, is depicted in Fig. 3. Different from other thermodynamic properties, the heat capacity increases as the temperature increases. In general, energy can be stored in translational, rotational and vibrational energy storage modes. An increase of the number of atoms in the cationic structure and consequently the number of contributing storage modes is behind the observed slightly larger isobaric heat capacity of [e2mpy][Tf2N] with respect to [epy][Tf2N] [18, 19]. In Fig. 4, the DSC curve for the ionic liquid is shown. During the heating process, [epy][Tf2N] shows two endothermic peaks at temperatures of 293 and 303 K, which indicates that there is a solid–solid transition and a melting process; these results are in excellent agreement with those of Liu et al. [9]. It has been observed that the melting point increases with a more symmetric cation [20]. The lack of a methyl group in [epy][Tf2N] makes it more symmetric and, consequently, it has the higher melting point. On cooling the ionic liquid, it showed an exothermic peak at T C = 268 K when the compound easily crystallized.

Isobaric molar heat capacities, C p , as a function of temperature

Thermal behavior around ambient temperature
Furthermore, two transport properties, electrical conductivity and dynamic viscosity, have been measured as a function of temperature, and these properties are plotted in Fig. 5. Their values were fitted to a Vogel–Fulcher–Tammann equation [21–23] since they present convex curves profiles:
where κ ∞, η 0, B and T 0 are adjustable parameters, and T is the absolute temperature. According to the Arrhenius equation, it may be deduced that the fitting parameters have physical meaning. On the one hand, κ ∞ and η 0 represent the maximum electrical conductivity and the minimum viscosity, respectively, if the temperature were infinite. On the other hand, the activation energy, E a, represents the energy needed for an ion to hop into a free hole [24] and it can be calculated from B = E a/k B, where k B is the Boltzmann constant. From these coefficients, the activation energies obtained are 0.0271 and 0.0656 eV for electrical conduction and viscosity, respectively.

The electrical conductivity increases rapidly with increasing temperature. Despite their ionic nature, ionic liquids have low conductivity due to the strong interactions among their ions. If we examine the results, it is remarkable that the behavior of this property depends on the size of the cation, an increase of the cation size causes a decrease of the conductivity. In this way, the absence of the methyl group in [epy][Tf2N] allows the ions in the ionic liquid to move well, thus enhancing transport within the fluid [25] and leads to higher κ values. With respect to the viscosity, the results demonstrate that the viscosity of [epy][Tf2N] decreases faster than electrical conductivity with increasing temperature. Viscosity depends strongly on the mobility and the intermolecular interactions among ions. The smaller viscosity values of [epy][Tf2N] compared with [e2mpy][Tf2N] are due to the fact that the cation without a methyl group is more symmetrical and therefore it moves better in the fluid. Also, the van der Waals and coulombic interactions are stronger in [e2mpy][Tf2N] [18, 26]. Both effects lead to a higher viscosity for the methylated ionic liquid. Finally, different studies involving the [Tf2N] anion demonstrated that ionic liquids with this anion show lower viscosities than those with other anions like [BF4]− or [TFO]− [27, 28].
Finally, for comparison, the data found in the literature were plotted in the corresponding figures. In general terms, the agreement of our values with published ones are good, with differences lower than 6 % in the worst case.
4 Theoretical Study
The number of theoretical studies of ionic liquids has grown in the last decade [29, 30]; theoretical methods have proved to be a powerful tool to investigate the structure and properties of individual ions and also the directionality and extent of the interaction between the cation and the anion that form a given ionic liquid. The information obtained from these studies is very useful for a better understanding of the thermophysical properties of ionic liquids and also for the design of novel ionic liquids.
With this aim, we have carried out DFT calculations to find different optimized geometries for the anion–cation pair [epy][Tf2N] using the Gaussian 03 package [31]. First, the individual ion’s structures were optimized separately, and then optimizations of the ionic pairs were performed by starting the calculations with five different possible orientations of the anion relative to the cation, as illustrated in Fig. 6. The level of theory was B3LYP/6-31+G(3df) and optimizations were carried out while ignoring symmetry. Vibration frequencies were calculated to confirm that true local minima of the PES are found in the calculations and the electronic energies were corrected using ZPEs (zero point vibrational energies).

Initial relative positions of [Tf2N]− around the [epy]+
Four local minima structures together with their relative energies are shown in Fig. 7. These structures correspond to four of the five starting geometries, while it has not been possible to find any local minimum when the anion is initially placed in front of the pyridinium ring.

B3LYP/6-31+G(3df) optimized geometries and relative interaction energies (in square brackets, units kJ·mol−1) for [epy][Tf2N]
The most stable structure found for the ionic pair [epy][Tf2N] is denoted as i in Fig. 7. In this configuration, the nitrogen and oxygen atoms of the anion are close to the nitrogen atom of the cation. The relative energy is −53.59 kJ·mol−1 and the distance between nitrogen atoms is 3.33 Å. Structure ii is obtained when the anion is placed aside the cation, oxygen atoms of the anion are close to two of the aromatic CH groups of the cation, it is less stabilizing than i, the relative energy is −49.47 kJ·mol−1 and the nitrogens are separated 5.24 Å. Structures iii and iv present the nitrogen atom of the anion close to one of the carbon atoms of the ethyl group of the cation, the distance between N atoms of the anion and cation are 4.24 and 5.50 Å, respectively, and the relative energies −43.93 and −43.49 kJ·mol−1.
To interpret the results obtained it should be noted that the acidic character of the aromatic hydrogen atoms is lower in pyridinium derivatives than in other aromatic cations such as imidazolium derivatives. The presence of only one nitrogen atom and the bigger size of the pyridinium ring also causes the interaction energies between ions to be lower in pyridinium-based ionic liquids [30, 32]. Moreover, Tsuzuki et al. [33] have shown that the nitrogen and oxygen atoms of the anion [Tf2N] have a large negative charge. Our previous studies for the ionic pairs [e2mpy][Tf2N] and [p2mpy][Tf2N] also show similar results [13], i.e. the structures with minimum energy correspond to geometries where the nitrogen and oxygen atoms of the anion are close to the nitrogen atom of the cation.
Density values of the bulk ionic liquid were estimated ab initio by simulation, in order to assess the predictive value of such a purely theoretical model for this class of compounds. Classical molecular simulations of the ionic liquid at different temperatures were performed using the GROMACS package [34]. The simulations were carried out on a NPT ensemble containing 200 anion–cation pairs, a total of 6,600 atoms, in a cubic box with three dimensional periodic boundary conditions, using a leap-frog integrator [35] of the equations of motion. Temperatures (spanning the interval from 283.15 to 338.15 K) were fixed by velocity rescaling (τ = 1 ps) [36]; the isotropic pressure was fixed at p = 1.01325 bar using a Berendsen-like algorithm [37] (τ p = 1 ps). Molecular and intermolecular interactions were described by the General Amber Force Field (GAFF) [38]; intermolecular interactions were computed with a cut-off radius of 1.0 nm, with long-range particle-mesh Ewald electrostatics [39]. After equilibration (density drift over 50 ns below ± 0.2 %), the dynamics were resolved over a production run of 50 × 106 steps with a step size of Δt = 0.001 ps, for a total simulation time of 50 ns. The system density was recorded every 10 ps, for a total of 5,000 data points per run, from which average and root-mean-square deviation density values were obtained. The distribution of densities over the 50 ns simulation run is correct, with a relative root-mean-square deviation of 0.249 %.
A graphical comparison between calculated densities and fitted experimental densities is shown in Fig. 8, while both calculated values of density at different temperatures together with the corresponding experimental values can be found in Table S2 of the Supporting Information. Although the simulation predictions overestimate the density for all points, they do so in a consistent fashion, reproducing the temperature trend with an average relative error of 1 %. The performance of this purely theoretical simulation prediction is thus satisfactory, comparable to that of other, non-computational predictive models. Further refinement of the force field and system size election have thus the potential of providing accurate predictions for this class of ionic liquids.

Comparison between calculated densities (open square) and fitted experimental densities (straight line)
Radial distribution functions were calculated for all the ions. In Fig. 9 the radial distribution functions for anion–anion, anion–cation and cation–cation interactions are shown. The distribution function of cations around anions or vice versa, in particular, can be used to gauge the average interionic distances and the degree of structural order of ionic pairs. Anion–cation radial distribution functions are remarkably consistent throughout the temperature range, with the population maximum at an intermolecular distance of ca. 0.77 nm for all cases. Interestingly, the cation–cation radial distribution functions depend on temperature, with two closest-neighbor population maxima at ca. 0.75 and ca. 1.05 nm. The relative population at these peaks changes with temperature, with higher temperatures favoring the bigger distance (1.05 nm). On the other hand, the anion–anion distribution functions present a single closest-neighbor maximum. This points to a non-trivial structure of the ionic liquid, with closely linked ionic pairs oriented so that cations occupy two non-equivalent environments.

Radial distribution functions, g (r), for [epy][Tf2N] at T = 308.15 K: anion–anion (black solid line); anion–cation (red dashed line); cation–cation (blue dashed line) (Color figure online)
5 Conclusions
In this paper, several thermophysical properties of n-ethylpyridinium bis(trifluoromethylsulfonyl)imide have been studied: density, speed of sound, refractive index, surface tension, isobaric molar heat capacity, electrical conductivity and kinematic viscosity have been measured in the temperature range 288.15–338.15 K. Moreover, from these experimental results, the isobaric expansibility, isentropic compressibility, molar refraction, entropy and enthalpy of surface per unit surface area, and dynamic viscosities have been obtained. Results were compared with those obtained previously for [e2mpy][Tf2N] to analyze the effect of methyl group on the pyridine cation. We have shown that the methyl group in the pyridine ring increases the values of speed of sound, refractive index, surface tension, viscosity and isobaric molar heat capacity. On the contrary, the density and ionic conductivity are higher for [epy][Tf2N].
With the aim of improving knowledge about these ionic liquids, we have also carried out a theoretical study to determine the relative energies of different structures of this compound. From these results, we have concluded that the structures with minimum energy correspond to geometries where the nitrogen and oxygen atoms of the anion are close to the nitrogen atom of the cation. Finally, using classical molecular dynamics simulations, the density and the structure of the ionic liquid were explored. The density dependence on temperature is consistent with the expected behavior, and the density predictions can be considered satisfactory.
References
Wasserscheid, P., Welton, T.: Ionic Liquids in Synthesis. Wiley, Weinheim (2003)
Ishikawa, M., Sugimoto, T., Kikuta, M., Ishiko, E., Kono, M.: Pure ionic liquid electrolytes compatible with a graphitized carbon negative electrode in rechargeable lithium-ion batteries. J. Power Sources 162, 658–662 (2006)
Gan, Q., Rooney, D., Xue, M., Thompson, G., Zou, Y.: An experimental study of gas transport and separation properties of ionic liquids supported on nanofiltration membranes. J. Membr. Sci. 280, 948–956 (2006)
Torimoto, T., Tsuda, T., Okazaki, K., Kuwabata, S.: New frontiers in materials science opened by ionic liquids. Adv. Mater. 22, 1196–1221 (2010)
Bermudez, M.-D., Jimenez, A.-E., Sanes, J., Carrion, F.-J.: Ionic liquids as advanced lubricant fluids. Molecules 14, 2888–2908 (2009)
Pereiro, A.B., Araujo, J.M.M., Esperanca, J.M.S.S., Marrucho, I.M., Rebelo, L.P.N.: Ionic liquids in separations of azeotropic systems—a review. J. Chem. Thermodyn. 46, 2–28 (2012)
Moniruzzaman, M., Nakashima, K., Kamiya, N., Goto, M.: Recent advances of enzymatic reactions in ionic liquids. Biochem. Eng. J. 48, 295–314 (2010)
Kato, R., Gmehling, J.: Activity coefficients at infinite dilution of various solutes in the ionic liquids [MMIM]+[CH3SO4]−, [MMIM]+[CH3OC2H4SO4]−, [MMIM]+[(CH3)2PO4]−, [C5H5NC2H5]+[(CF3SO2)(2)N]− and [C5H5NH]+[C2H5OC2H4OSO3]−. Fluid Phase Equilib. 226, 37–44 (2004)
Liu, Q.-S., Yang, M., Yan, P.-F., Liu, X.-M., Tan, Z.-C., Welz-Biermann, U.: Density and surface tension of ionic liquids [C(n)py][NTf2] (n = 2, 4, 5). J. Chem. Eng. Data 55, 4928–4930 (2010)
Zhang, Q.-G., Sun, S–.S., Pitula, S., Liu, Q.-S., Welz-Biermann, U., Zhang, J.-J.: Electrical conductivity of solutions of ionic liquids with methanol, ethanol, acetonitrile, and propylene carbonate. J. Chem. Eng. Data 56, 4659–4664 (2011)
Bittner, B., Wrobel, R.J., Milchert, E.: Physical properties of pyridinium ionic liquids. J. Chem. Thermodyn. 55, 159–165 (2012)
Gutmann, T., Sellin, M., Breitzke, H., Stark, A., Buntkowsky, G.: Para-hydrogen induced polarization in homogeneous phase—an example of how ionic liquids affect homogenization and thus activation of catalysts. Phys. Chem. Chem. Phys. 11, 9170–9175 (2009)
Garcia-Mardones, M., Bandres, I., Carmen Lopez, M., Gascon, I., Lafuente, C.: Experimental and theoretical study of two pyridinium-based ionic liquids. J. Solut. Chem. 41, 1836–1852 (2012)
Mokhtarani, B., Sharifi, A., Mortaheb, H.R., Mirzaei, M., Mafi, M., Sadeghian, F.: Density and viscosity of pyridinium-based ionic liquids and their binary mixtures with water at several temperatures. J. Chem. Thermodyn. 41, 323–329 (2009)
Gu, Z.Y., Brennecke, J.F.: Volume expansivities and isothermal compressibilities of imidazolium and pyridinium-based ionic liquids. J. Chem. Eng. Data 47, 339–345 (2002)
Matkowska, D., Goldon, A., Hofman, T.: Densities, excess volumes, isobaric expansivities, and isothermal compressibilities of the 1-ethyl-3-methylimidazolium ethylsulfate plus ethanol system at temperatures (283.15 to 343.15 K) and pressures from 0.1 to 35 MPa. J. Chem. Eng. Data 55, 685–693 (2010)
Carvalho, P.J., Freire, M.G., Marrucho, I.M., Queimada, A.J., Coutinho, J.A.P.: Surface tensions for the 1-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ionic liquids. J. Chem. Eng. Data 53, 1346–1350 (2008)
Crosthwaite, J.M., Muldoon, M.J., Dixon, J.K., Anderson, J.L., Brennecke, J.F.: Phase transition and decomposition temperatures, heat capacities and viscosities of pyridinium ionic liquids. J. Chem. Thermodyn. 37, 559–568 (2005)
Liu, H., Maginn, E., Visser, A.E., Bridges, N.J., Fox, E.B.: Thermal and transport properties of six ionic liquids: an experimental and molecular dynamics study. Ind. Eng. Chem. Res. 51, 7242–7254 (2012)
Ngo, H.L., LeCompte, K., Hargens, L., McEwen, A.B.: Thermal properties of imidazolium ionic liquids. Thermochim. Acta 357, 97–102 (2000)
Vogel, H.: Das Temperatur-abhängigkeitsgesetz der viskosität von flüssigkeiten (The temperature-independence law of viscosity of liquids). Phys. Zeit. 22, 645–646 (1921)
Tammann, G., Hesse, W.: Die Abhängigkeit der viscosität von der temperatur bei unterkühlten flüssigkeiten (The temperature dependence of the viscosity of supercooled fluids). Zeitschrift für anorganische und allgemeine Chemie 156, 245–257 (1926)
Fulcher, G.S.: Analysis of recent measurements of the viscosity of glasses. J. Am. Ceram. Soc. 8, 339–355 (1923)
Yu, Y.-H., Soriano, A.N., Li, M.-H.: Heat capacities and electrical conductivities of 1-n-butyl-3-methylimidazolium-based ionic liquids. Thermochim. Acta 482, 42–48 (2009)
Vila, J., Varela, L.M., Cabeza, O.: Cation and anion sizes influence in the temperature dependence of the electrical conductivity in nine imidazolium based ionic liquids. Electrochim. Acta 52, 7413–7417 (2007)
Gardas, R.L., Coutinho, J.A.P.: A group contribution method for viscosity estimation of ionic liquids. Fluid Phase Equilib. 266, 195–201 (2008)
Fredlake, C.P., Crosthwaite, J.M., Hert, D.G., Aki, S., Brennecke, J.F.: Thermophysical properties of imidazolium-based ionic liquids. J. Chem. Eng. Data 49, 954–964 (2004)
Seoane, R.G., Corderi, S., Gomez, E., Calvar, N., Gonzalez, E.J., Macedo, E.A., Dominguez, A.: Temperature dependence and structural influence on the thermophysical properties of eleven commercial ionic liquids. Ind. Eng. Chem. Res. 51, 2492–2504 (2012)
Izgorodina, E.I.: Towards large-scale, fully ab initio calculations of ionic liquids. Phys. Chem. Chem. Phys. 13, 4189–4207 (2011)
Rees, R.J., Lane, G.H., Hollenkamp, A.F., Best, A.S.: Predicting properties of new ionic liquids: density functional theory and experimental studies of tetra-alkylammonium salts of (thio)carboxylate anions, RCO2 −, RCOS− and RCS2 −. Phys. Chem. Chem. Phys. 13, 10729–10740 (2011)
Frisch, G.W.T.M.J., Pople, J.A., Schlegel, H.B., Scuseria, G.E., Robb, M.A., Cheeseman, J.R., Montgomery Jr., J.A., Vreven, T., Kudin, K.N., Burant, J.C., Millam, J.M., Iyengar, S.S., Tomasi, J., Barone, V., Mennucci, B., Cossi, M., Scalmani, G., Rega, N., Petersson, G.A., Nakatsuji, H., Hada, M., Ehara, M., Toyota, K., Fukuda, R., Hasegawa, J., Ishida, M., Nakajima, T., Honda, Y., Kitao, O., Nakai, H., Klene, M., Li, X., Knox, J.E., Hratchian, H.P., Cross, J.B., Bakken, V., Adamo, C., Jaramillo, J., Gomperts, R., Stratmann, R.E., Yazyev, O., Austin, A.J., Cammi, R., Pomelli, C., Ochterski, J.W., Ayala, P.Y., Morokuma, K., Voth, G.A., Salvador, P., Dannenberg, J.J., Zakrzewski, V.G., Dapprich, S., Daniels, A.D., Strain, M.C., Farkas, O., Malick, D.K., Rabuck, A.D., Raghavachari, K., Foresman, J.B., Ortiz, J.V., Cui, Q., Baboul, A.G., Clifford, S., Cioslowski, J., Stefanov, B.B., Liu, G., Liashenko, A., Piskorz, P., Komaromi, I., Martin, R.L., Fox, D.J., Keith, T., Al-Laham, M.A., Peng, C.Y., Nanayakkara, A., Challacombe, M., Gill, P.M.W., Johnson, B., Chen, W., Wong, M.W., Gonzalez, C.: Gaussian. Gaussian Inc., Wallingford, CT (2004)
Xuan, X., Guo, M., Pei, Y., Zheng, Y.: Theoretical study on cation–anion interaction and vibrational spectra of 1-allyl-3-methylimidazolium-based ionic liquids. Spectrochim. Acta A 78, 1492–1499 (2011)
Tsuzuki, S., Tokuda, H., Hayamizu, K., Watanabe, M.: Magnitude and directionality of interaction in ion pairs of ionic liquids: relationship with ionic conductivity. J. Phys. Chem. B. 109, 16474–16481 (2005)
Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E.: GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435–447 (2008)
Hockney, R., Goel, S., Eastwood, J.: Quiet high-resolution computer models of a plasma. J. Comput. Phys. 14, 148–158 (1974)
Bussi, G., Donadio, D., Parrinello, M.: Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007)
Berendsen, H., Postma, J., Vangunsteren, W., Dinola, A., Haak, J.: Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984)
Wang, J.M., Wolf, R.M., Caldwell, J.W., Kollman, P.A., Case, D.A.: Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004)
Essmann, U., Perera, L., Berkowitz, M., Darden, T., Lee, H., Pedersen, L.: A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 (1995)
Acknowledgments
The authors gratefully acknowledge financial support from Diputación General de Aragón and Fondo Social Europeo “Construyendo Europa desde Aragón”.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Benito, J., García-Mardones, M., Pérez-Gregorio, V. et al. Physicochemical Study of n-Ethylpyridinium bis(trifluoromethylsulfonyl)imide Ionic Liquid. J Solution Chem 43, 696–710 (2014). https://doi.org/10.1007/s10953-014-0156-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10953-014-0156-5