
Abstract
Pure and erbium-doped ZnO nanoparticles (Zn1−xErxO), (0.00 ≤ x ≤ 0.10), were synthesized by wet chemical co-precipitation method. The structural, optical, and magnetic properties of the prepared samples were investigated using x-ray powder diffraction (XRD), transmission electron microscopy (TEM), ultraviolet-visible spectroscopy (UV), Fourier transform infrared spectroscopy (FTIR), M-H magnetic hysteresis, and electron paramagnetic resonance (EPR). XRD studies exhibit the presence of a single ZnO wurtzite hexagonal crystal structure for 0.00 ≤ x ≤ 0.06. A secondary phase of Er2O3 appears for x > 0.06. This means that the solubility limit for doping Zn2+ ions by Er3+ ions is about x = 0.06 under our preparations condition. The lattice parameter a is not affected by the Erbium doping. On the other hand, the lattice parameter c and the unit cell volume V increase with the increase of x up to x = 0.06. This is attributed to the larger ionic size of Er3+ ions (0.88 Å) compared to Zn2+ ions (0.74 Å). Both c and V decrease for x > 0.06. TEM micrographs indicate that the shape and the size of the ZnO nanoparticles are modified by changing the doping level of Er. The UV measurements point out that band gap energy Eg decreases with the increase of x up to x = 0.06. Then, it increases for both x = 0.08 and 0.10. FTIR spectra confirm the presence of O–H and Zn–O stretching modes at 3451.963 and 428.901 cm− 1, respectively, in pure and doped ZnO samples. The Zn–O stretching mode shifts toward a lower wavenumber for x = 0.06 and toward a higher wavenumber for x = 0.10. M-H hysteresis analysis, at room temperature, reveals that the pure ZnO has a ferromagnetic signal combined with diamagnetic and paramagnetic contributions. This ferromagnetism is reduced for the doped samples up to x = 0.02, and an antiferromagnetic alignment appears for 0.04 ≤ x ≤ 0.10. The saturation magnetization (Ms), the coercivity (Hc), the retentivity (Mr), the anisotropy constant (Ka), and the magnetic moment (μm) were estimated and discussed in terms of erbium doping for the different samples. EPR spectra for Zn1−xErxO were measured at room temperature in order to study the effect of Er substitution on the g value, resonance field (Hr), peak to peak line width (ΔHpp) and spin–spin relaxation time constant (T2).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
ZnO is an n-type semiconductor that is characterized by a large exciton binding energy of 60 meV at room temperature, a direct band gap of 3.37 eV, and a high transmittance in visible region [1]. These distinctive properties have received much attention for their use in light-emitting diodes (LED) [2], solar cells [3], spintronic devices [4], photonic catalysis [5], transparent conductors [6], and gas sensors [7]. The doping of ZnO by transition [8, 9] and rare earth elements [10, 11] is an interesting topic in the last few years because it has pronounced effect on structural, optical, electrical, and magnetic properties of ZnO. Sharrouf et al. [9] studied the effect of Mn doping on ZnO nanoparticles, preparing by co-precipitation technique. They found that the doped samples exhibit a decrease in band gap energy (Eg) with increasing x. Moreover, doping ZnO with Mn at different concentrations results in modification of the magnetic properties for the doped samples. Samples with 0.01 ≤ x ≤ 0.03 revealed a ferromagnetic behavior, while samples with 0.05 ≤ x ≤ 0.10 combined ferromagnetic and paramagnetic properties. Liu et al. [12] investigated the effects of vanadium (V) doping concentrations on the structural, magnetic, and optical properties of Zn1−xVxO nanoparticles, synthesized by a sol-gel method. They found that samples with x ≤ 0.03 have room-temperature ferromagnetism. Based on the results of Raman and photoluminescence, they concluded that oxygen vacancies (Vo) are the origin of the ferromagnetism in Zn1−xVxO. Additionally, the increase of V-doping concentration caused an increase in the values of the saturation magnetization (Ms). Yadav et al. [13] synthesized pure and 5 M % Eu-doped ZnO nanorods using a wet-chemical precipitation method. The Eu-doped ZnO sample showed a higher dielectric constant and ferroelectric phase transition at 230 °C. The Eu doping in ZnO increased the leakage current and the DC conductivity while the knee voltage decreased making Eu-doped ZnO a useful material for solar cell applications. Porkalai et al. [14] used the sol-gel method to prepare pure and Lanthanum doped ZnO nano particles. They found that doping with Lanthanum enhanced the luminescence and increased the current conductivity of the doped samples. Likewise, optical band gap increased from 3.28 eV to 3.44 eV as a result of increasing La-doping concentration. Oprea et al. [15] explored the effect of Gd(III) ions doping on the photoluminescence, magnetic properties and photocatalytic activity of ZnO nanoparticles prepared by simple sol-gel method. The increase of Gd doping concentrations in the ZnO led to a drop in the near band edge position, improvement of the blue-green luminescence, and a better photocatalytic activity. The saturation magnetization (Ms) per Gd(III) ion decreased with increasing Gd content in ZnO. This result was attributed to the antiferromagnetic alignment caused by the increased number of Gd atoms. Pradyumnan et al. [16] investigated the thermoelectric properties in terms of electrical and thermal transport in pure and holmium-doped ZnO compounds prepared by solid-state reaction method. Doping Ho in ZnO decreased the resistivity while it enhanced the power factor making the Ho-doped samples suitable in thermoelectric applications. The effects of erbium doping on ZnO nanosheets synthesized by the hydrothermal and post-annealing techniques were examined by Lang et al. [11]. The photoluminescence (PL) spectra exhibited both ultraviolet and deep-level emission. The intensity of the whole deep-level emission was improved with increasing erbium concentration. Furthermore, erbium dopants produced more defects leading to a worse crystallization of the doped nanosheets. In this work, we studied structural, optical, and magnetic properties of Zn1−xErxO (0.00 ≤ x ≤ 0.10) prepared by co-precipitation technique. For this investigation, the prepared samples were investigated using x-ray powder diffraction (XRD), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FTIR), UV-Visible spectroscopy, M-H hysteresis analysis, and Electron paramagnetic resonance (EPR).
2 Experimental Techniques
2.1 Preparation of Nano Powder
The chemical substances used in the present co-precipitation technique were zinc chloride (ZnCl2) and erbium (III) chloride hexahydrate (ErCl3.6H2O). Based on stoichiometric ratios, the amounts of each substance were calculated by changing x from 0.00 up to 0.10 in Zn1−xErxO nanoparticles. Substances were dissolved in distilled water, mixed and stirred with a magnetic stirrer, and 4.0 M sodium hydroxide solution (NaOH) was added dropwise until pH = 12, approximately. The obtained solution is then heated at 60 °C for 2 h with continuous and constant stirring rate. The product was washed with distilled water to reach a pH around 7, then dried at 100°C for 18 h to remove water, and finally kept at 550 °C for 4 h in order to improve the crystallinity of the prepared samples.
2.2 Characterization of Nano Powder
The obtained nano-powders of Zn1−xErxO were characterized by x-ray powder diffraction measurements at room temperature using Bruker D8 advance powder diffractometer with Cu-Kα radiation (λ = 1.54056 Å) in the range of 25°≤ 2𝜃 ≤ 75°. TEM micrographs were obtained by using Jeol transmission electron microscope JEM-100CX, operated at 80 kV. The FTIR spectra of the powder samples were recorded using FTIR 8400S Shimadzu spectrometer. UV-Visible measurements were performed using the ultraviolet-visible-near infrared (NIR) spectrophotometer V-670 that records the absorption spectra at a wavelength of 190–2500 nm at room temperature. Magnetic measurements were carried out at room temperature using a vibrating sample magnetometer (Lake Shore 7410) having temperature range capability from 4.2 to 1273 . EPR measurements of powder samples were conducted using Bruker Elexsys 500 EPR spectrometer.
3 Results and Discussion
3.1 Structure and Morphology
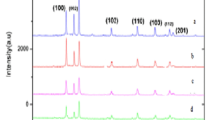
XRD patterns of pure and Er-doped ZnO nanoparticles are displayed in Fig. 1. The planes (100), (002), (101), (102), (110), (103), (200), (112), and (201) correspond to the hexagonal (wurtzite) structure of ZnO. The Er2O3 peaks are not observed up to x = 0.06, suggesting that there is a complete substitution of erbium in the zinc sites. For x > 0.06, Er2O3 peaks appear in addition to wurtzite phase of ZnO. The lattice parameters (a and c) and the unit cell volume (V ) of Zn1−xErxO nanoparticles are calculated using the following equations:
where d is the interplanar distance and (h, k, l) are the Miller indices. Lattice parameters values, the ratio c/a and the unit cell volume (V ) for pure and Er-doped samples are listed in Table 1. It can be noticed that a is not affected by erbium doping since a = 3.243 ± 0.004 Å. While the lattice parameter (c) and the unit cell volume (V ) show an increase in their values up to x = 0.06 with respect to the pure sample. This behavior of c and V may be attributed to the larger ionic size of Er3+(0.88 Å) substituting Zn2+ (0.74 Å) in ZnO lattice. Both c and V decrease for x > 0.06 and reach the value of pure ZnO. This cell contraction may be explained such as the larger Er3+ ions leave the ZnO lattice at high doping concentrations (x > 0.06) to form Er2O3 impurity [17, 18]. The decrease in lattice parameter c may also be attributed to the formation of oxygen vacancies in ZnO lattice as a result of increasing the number of dopant (Er3+) ions as it was reported by Dakhel et al. [19] for Gd3+-doped ZnO powder prepared by co-precipitation method. The ratio (c/a) is 1.60 for all samples. This value is very close to the ideal value for hexagonal cell (c/a = 1.633) [20]. The average crystallite sizes are estimated using Scherrer equation:

XRD patterns recorded from undoped and Er3+ doped ZnO
where D is the average crystallite size in nanometers, K is a constant or shape factor and equals to 0.9, λ is the wavelength of the x-ray radiation, βhkl is the peak width at half maximum intensity, and 𝜃 is the peak position. The values of D are tabulated in Table 1. The average crystallite size of Zn1−xErx O nanoparticles decreases with increasing erbium content up to x = 0.04 and beyond x = 0.04, and the crystallite size increases to a max value of 50.3 nm among all the prepared samples for x = 0.06. Similar trends of variation of crystallite size were also observed by Divya et al. [21] for erbium-doped ZnO crystal prepared by solid-state reaction method. They attributed the initial decrease of the crystallite size to the deformation of the ZnO lattice by larger Er3+ ions leading to a lowering of nucleation and decreasing growth rate of ZnO crystals. While the increase in crystallite size may imply that erbium ions were dissolved in ZnO grains and the excess of Er3+ head toward the grain boundaries and enhances the liquid-phase sintering. For x = 0.08 and x = 0.10, the crystallite size decreases again for the same reasons mentioned above.
The shape and the size of pure and erbium-doped ZnO nanoparticles were investigated using the TEM technique and the TEM images are shown in Fig. 2a–d for x = 0.00, 0.02, 0.04, and 0.10, respectively. The particle sizes were measured and found to be in the range of 22.9–36 nm, which are in good agreement with the crystallite sizes calculated from XRD analysis. The particles have no definite shape for samples with x = 0.00 and 0.10. This finding is consistent with the results obtained by Vaseem et al. [22] since the reaction is carried out in dry air, the produced ZnO nanoparticles have a lack of defined shape or size. Moreover, the absence of definite shape may reflect that damages in recrystallization of ZnO lattice occurred because of the high-temperature heating process [21]. The shape of the particles changes to nano-like rods for 0.01 ≤ x ≤ 0.08, which can be due to the erbium doping level that modifies the shape of the nanoparticles [9].

TEM images for Zn1−xErxO with ax = 0.00, bx = 0.02, cx = 0.04, and dx = 0.10
4 FTIR Analysis
FTIR spectra of Zn1−xErxO nanoparticles (x = 0.00, 0.06, and 0.10) between 400 and 4250 cm− 1 at room temperature are shown in Fig. 3. The peaks at 3451.963 and 1116.503 cm− 1 correspond to O–H stretching and deformation, respectively, assigned to the water adsorption on the metal surface [23]. The peaks at 428.901 and 1635.683 cm− 1 could be attributed to the ZnO stretching mode [23, 24]. It can be noticed that the peak at 428.901 cm− 1, appearing in the spectrum of the pure ZnO sample, is shifted a little to a lower wavenumber for x = 0.06 and to a higher wavenumber for x = 0.10 with a change in the percentage of transmittance for the doped samples compared to the pure sample as shown in the inset of Fig. 3 .This can prove that the presence of Er causes a perturbation in the surrounding of Zn–O–Zn network. The small shift in Zn–O stretching peak may be caused by the change in the parameters and the bond properties of Zn perturbed by Er doping [9]. A peak at 1055.990 cm− 1 appears in pure ZnO which may correspond to the alcoholic C–O stretching mode, but this peak disappears for the doped samples. In addition, bands appearing between 750 and 1020 cm− 1 (886.552 and 987.408 cm− 1) in doped ZnO samples may correspond to the bending and twisting vibration of ZnOH. The absorption peaks in the range of 1100–1600 cm− 1 appearing in the pure and doped samples could be attributed to the Zn–OH bending mode, to C–OH plane bending and C–OH out-of-plane bending [25].

FTIR spectra of Zn1−xErxO nanoparticles (x = 0.00, 0.06, and 0.10). The inset shows the absorption peaks of Zn1−xErxO for x = 0.00, 0.06, and 0.10 at Zn–O stretching vibration
4.1 UV-Spectroscopy
UV-visible spectra of Zn1−xErxO (x = 0.00, 0.01, 0.02, 0.04, and 0.08) are shown in Fig. 4. A characteristic absorption peak of wurtzite hexagonal pure ZnO appears in the spectrum of pure sample at 377 nm. The absorption peaks of doped samples are blue shifted towards smaller wavelengths in comparison with the peak of pure sample. This shift may reflect that Er3+ ions are well integrated in ZnO lattice [26]. The energy band gaps (Eg) for allowed direct transitions of Zn1−xErxO are calculated using Tauc relation:
α is the absorption coefficient, B is a constant, hν is the energy of incident photons, and n = 1/2 for allowed direct transitions. Eg is obtained from the plot of (αhν)2 versus hν, then the curve is extrapolated to (αhν)2 = 0 as shown in Fig. 5a, b for x = 0.00 and 0.10. The obtained values of Eg are listed in Table 2. Eg decreases with increasing erbium doping up to x = 0.06 then it increases for x = 0.08 and 0.1. The drop in Eg may be due to the erbium electron-localized states that create new states near to the conduction band, leading to a reduction in the band gap of pure ZnO [27]. The variation of band gap depends also on particle size and lattice parameters. The decrease in particle size with increasing doping concentration leads to a rising of the band gap as the quantum refinement effect occurs [10]. The Bras’ effective mass model [28] can also explain our results. The energy gap (Eg) of the nanoparticles can be expressed, according to Bras’ effective mass model, as a function of particle size, as given by the following expression:
where Eg is the measured band gap of the nanoparticles, Ebulk is the band gap of the bulk semiconductor, \( \hbar \) is the second Plank’s constant, R is the radius of the nanoparticle, me is the effective mass of the electron, mh is the effective mass of the hole, e is the charge of electron, and ε is the electric permittivity of the material. It is clear from (5) that the band gap increased as particle size decreased.

The UV-Visible spectroscopy of Zn1−xErxO for x = 0.00, 0.01, 0.02, 0.04, and 0.08

(αhν)2 versus photon energy (hν) of Zn1−xErxO for ax = 0.00 and bx = 0.10
4.2 Magnetic Measurements
Magnetic hysteresis of Zn1−xErxO nanoparticles were recorded at room temperature and the results are plotted in Fig. 6a–d for x = 0.00, 0.01, 0.02, and 0.10, respectively. The pure ZnO nanoparticles exhibit room-temperature ferromagnetism (RTFM) behavior which may be attributed to some defects at the surface, interface, and grain boundary [29] or to lattice defects such as oxygen vacancy (Vo) or zinc interstitial (Zni) in pure ZnO nanoparticles [30]. Moreover, the hysteresis loop of pure ZnO sample also shows a diamagnetic and paramagnetic contributions, those appear at about 0.8 Tesla as M decreases with the increasing of H. These contributions were also observed in ZnO nanocrystals capped with polyvinyl pyrrolidone (PVP), synthesized using the wet chemical route at room temperature by Phadnis et al. [31]. The presence of both paramagnetic and diamagnetic contributions combined with ferromagnetic contribution indicates the surface and ligand-induced defects [32], confirming the TEM results. The sample with x = 0.01 also shows an RTFM which may be due to the substitution of Er3+ in Zn2+ sites rather due to the formation of any magnetic secondary phases. Similar results were obtained for Er-doped ZnO thin films prepared by coupled plasma enhanced physical vapor deposition [33]. The observed ferromagnetic behavior in the doped sample (x = 0.01) can also be explained by the bound magnetic polaron (BMP) model [34]. The weakening of ferromagnetism (FM) for x = 0.02 may be due to the fact that erbium exists near surface site because of non-consistency of ionic radii [32]. For the samples with 0.04 ≤ x ≤ 0.10, the anti-ferromagnetic (AFM) alignment is predominant. This phenomenon can be explained as follows: At high doping concentration of erbium, oxygen vacancies increase, leading to quenching of surface defects or shallow donors which promote AFM alignment and thus weaken the FM behavior [35]. The saturation magnetization (Ms) was measured directly from M-H curves for 0.00 ≤ x ≤ 0.02, and calculated for 0.04 ≤ x ≤ 0.10 by plotting M (emu/g) versus (1/H), as shown in Fig. 7 for x = 0.10. The values of anisotropy constant (Ka) were calculated from the following equation [36]:
where Hc is the coercivity (obtained from the hysteresis loops for each sample).
The magnetic moment (μm) per unit formula in Bohr magnetron is calculated from saturation magnetization by using the following formula:
where μm = magnetic moment, Mw = molecular weight, and 5585 = magnetic factor

M-H hysteresis loop of Zn1−xErxO nanoparticles for ax = 0.00, bx = 0.01, cx = 0.02, and dx = 0.10

The M versus 1/H curve for x = 0.10 extrapolated to 1/H = 0
The values of saturation magnetization (Ms), retentivity (Mr) (obtained from the hysteresis loops), coercivity (Hc), squareness ratio S = (Mr/Ms) [37], anisotropy constant (Ka), and the magnetic moment (μm) are tabulated in Table 3. It can be noticed that the saturation magnetization, the retentivity, and the magnetic moment have the same variation trends with the doping concentrations. For 0.00 ≤ x ≤ 0.02, both the saturation magnetization (Ms) and the magnetic moment (μm) increase. This result may be due to the defects such as oxygen vacancies and interstitials as it was reported by Kung et al. [38] for Y-doped ZnO nanoparticles. Upon further increase in Er-doping concentration, Ms, and μm follow a nearly decrease trend, which may be due to the antiferromagnetic ordering caused by the increasing numbers of erbium ions [15]. The Hc of Er3+-doped zinc oxide shows almost a decreasing trend. This is probably due to the decrease in the non-magnetic zinc cations at the expense of an increase in Er3+ content resulting in a lower anisotropy [39]. The squareness ratio is a measure of how square the hysteresis loop is. It decreases with increasing doping concentration. The values of S are lower than 0.5, indicating that the particles interact by magnetostatic interaction and the anisotropy decreases in crystal lattice [40, 41]. The values of anisotropy constant (Ka) display a nonlinear variation. This behavior may be attributed to the decrease in particle size, leading to a spin disorder in surface layer and resulting in reduced anisotropy [36]. EPR spectral analysis is an effective and powerful technique used to elucidate the electron spin dynamics and the defect centers in ZnO [42, 43]. Figure 8a–d show first-derivative EPR spectra of Zn1−xErxO nanoparticles for x = 0.00, 0.04, 0.08, and 0.10, recorded at room temperature. A clear ferromagnetic signal centered at a magnetic field of about 3486 (G) approximately and a paramagnetic signal at 3571.85 (G) are observed in the EPR spectrum of pure ZnO sample which confirm the results of the magnetic hysteresis loop (Fig. 6a). The EPR parameters such as g value, resonance field (Hr), peak to peak line width (ΔHpp), and spin–spin relaxation time constant (T2) were calculated, and the values are listed in Table 4. The relaxation times characterize the interaction of electron spins with the surrounding spins and within the lattice. The spin–spin relaxation time constant (T2) is determined from the following equations [44, 45]:
where β is Bohr magnetron and ΔH1/2 is the line width at half of the absorption peak. The variation of these parameters are controlled by two main factors, which are magnetic dipole interactions among nanoparticles and super-exchange interactions between the magnetic ions through oxygen ions [45]. In our work, the g values of pure ZnO and erbium-doped ZnO samples are either equal to ∼ 1.95 or ∼ 1.96 without displaying a significant trend of variation with increasing erbium content concentrations. The signal at g = 1.96 was also observed in pure ZnO nanoparticles calcinated at 500° [46] and it was attributed to oxygen vacancies [47,48,49]. Similar EPR signal with g = 1.96 was recorded in EPR spectrum of 2 wt.% of Er-doped ZnO nanoparticles prepared by co-precipitation method [35]. It was assigned to shallow donors (zinc interstitials, Zni) [50, 51]. The appearance of shallow donor level induced by the substitution of Er3+ in ZnO lattice is clearly demonstrated [52] by the signal at g∼ 1.95 appearing in the EPR spectrum of erbium-doped samples. Furthermore, Reddy et al. [26] observed sharp resonance signals at g = 1.994 and 2.007 in Eu-doped ZnO samples synthesized by auto ignition-based low-temperature solution combustion method in addition to other resonance signals. They revealed that these two values are close to free electron value (g = 2.0023), corresponding to an unpaired electron trapped on an oxygen vacancy site [53, 54]. Clearly, it is observed from Table 4 that the resonant field (Hr) slightly decreases with increasing doping concentrations. This drop can be explained as follows: as the erbium content increases, the neighboring Er3+ ions become close to each other leading to an enhancement of the super-exchange interactions between them. The improvement of the super-exchange interactions raises the internal field [55] and decreases the resonant field with increasing erbium concentration [56]. From Fig. 8b–d, we remark that the ferromagnetic signal disappears in doped samples indicating the weakening of the ferromagnetic behavior and the appearance of the AFM alignment due the increase of the erbium content This result is confirmed in the magnetic hysteresis loops of the samples. The most important change in the EPR signal with increasing doping content is the approximate increase of the line width leading to a broadening of the signals which is attributed to the dipolar interactions between the increasing number of dopant ions [57]. For 0.00 ≤ x ≤ 0.10, the approximate decrease in relaxation time is attributed to the decrease in electron motion, weakening of super exchange interaction in lattice [58], or to the decrease in the particle size [59]. This result is in agreement with the XRD analysis showing a nearly decreasing particle size with increasing erbium content.

EPR spectra of Zn1−xErxO for ax = 0.00, bx = 0.04, cx = 0.08 and dx = 0.10
5 Conclusion
Zn1−xErxO nanoparticles, 0.00 ≤ x ≤ 0.10, were synthesized by co-precipitation method. XRD analysis indicated the formation of hexagonal wurtzite structure of ZnO with erbium oxide secondary phase appearing for x > 0.06. TEM technique showed a nano-like rod shape for 0.01 ≤ x ≤ 0.08 and no definite shape was observed for pure ZnO and x = 0.10 sample. The formation of ZnO phase was confirmed by the FTIR analysis showing two major peaks: O–H stretching peak at 3451.963 cm− 1 and Zn–O stretching peak at 428.901 cm− 1 with a shift in the Zn–O peak due to the change of parameters and bond properties of ZnO lattice perturbed by Erbium doping. The band gap energies (Eg) calculated from the recorded UV-VIS spectra decreased for 0.00 ≤ x ≤ 0.06, then increased for x = 0.08 and 0.10. The M-H hysteresis loops at room temperature exhibited a room temperature ferromagnetic behavior for 0.00 ≤ x ≤ 0.02 while the antiferromagnetic behavior dominated for x ≥ 0.04 due to the formation of oxygen vacancies leading to a quenching of surface defects. A clear ferromagnetic signal combined with a paramagnetic signal were observed in the EPR spectrum of pure ZnO. The decrease in resonance field (Hr) and the broadening of the signal of the EPR measurements with increasing doping concentration proved the antiferromagnetic behavior of the samples with x ≥ 0.04.
References
Janotti, A., Van de Walle, C.G.: Fundamentals of zinc oxide as a semiconductor. Rep. Prog. Phys. 72, 126501 (2009)
Fan, J.C., Chang, S.L., Xie, Z.: devices ZnO-based light-emitting diodes. In: Pyshkin, S (ed.) Optoelectronics—advanced materials, pp. 25–58. InTech (2013)
Oku, T., Yamada, T., Fujimoto, K., Akiyama, T.: Microstructures and photovoltaic properties of Zn(Al)O/Cu2O-based solar cells prepared by spin-coating and electrodeposition. Coatings 4, 203–213 (2014)
Wolf, S.A., Awschalom, D.D., Buhrman, R.A., Daughton, J.M., von Molnar, S., Roukes, M.L., Chtchelkanova, A.Y., Treger, D.M.: Spintronics: a spin-based electronics vision for the future. Science 294, 1488–1495 (2001)
Behnajady, M.A., Modirshahla, N., Shokri, M., Zeininezhad, A., Zamani, H.A.: Enhancement photocatalytic activity of ZnO nanoparticles by silver doping with optimization of photodeposition method parameters. J. Environ. Sci. Health: Part A 44, 666–672 (2009)
Klingshirn, C.: ZnO: Material, physics and applications. Chem. Phys. Chem. 8(6), 782–803 (2007)
Wan, Q., Li, Q.H., Chen, Y.J., Wang, T.H., He, X.L., Li, J.P., Lin, C.L.: Fabrication and ethanol sensing characteristics of ZnO nanowire gas sensors. Appl. Phys. Lett. 84, 3654–3656 (2004)
Djaja, N.F., Montja, D.A., Saleh, R.: The effect of Co incorporation into ZnO nanoparticles. Adv. Mater. Phys. Chem. 3, 33–41 (2013)
Sharrouf, M., Awad, R., Marhaba, S., El-Said Bakeer, D.: Structural, optical and room temperature magnetic study of Mn-doped ZnO nanoparticles. NANO: Brief Reports Rev. 11, 1650042 (2016)
He, H.Y., Fei, J., Lu, J.: Sm-doping effect on optical and electrical properties of ZnO films. J. Nanostruct. Chem. 5, 169–175 (2015)
Lang, J., Wang, J., Zhang, Q., Xu, S., Han, D., Yang, J., Han, Q., Yang, L., Sui, Y., Li, X., Liu, X.: Synthesis and photoluminescence characterizations of the Er3+-doped ZnO nanosheets with irregular porous microstructure. Mater. Sci. Semicond. Process. 41, 32–37 (2016)
Liu, H., Liu, Y., Yang, L., Chen, Z., Liu, H., Li, W., Yang, J., Zhou, Z.: Role of oxygen vacancies in V-doped ZnO diluted magnetic semiconductors. J. Mater. Sci: Mater Electron. 26, 2466–2470 (2015)
Yadav, H., Sinha, N., Goel, S., Kumar, B.: Eu-doped ZnO nanoparticles for dielectric, ferroelectric and piezoelectric applications. J. Alloys Compd. 689, 333–341 (2016)
Porkalai, V., Benny Anburaj, D., Sathya, B., Nedunchezhian, G., Meenambika, R.: Study on the synthesis, structural, optical and electrical properties of ZnO and lanthanum doped ZnO nano particles by sol-gel method. Mech. Mater. Sci. Eng. J. 9 (2017)
Oprea, O., Vasile, O.R., Voicu, G., Craciun, L., Andronescu, E.: Photoluminescence, magnetic properties and photocatalytic activity of Gd3+ doped ZnO nanoparticle. Digest J. Nanomater. Biostruct. 7, 1757–1766 (2012)
Pradyumnan, P.P., Paulson, A., Muhammed Sabeer, N.A., Deepthy, N.: Enhanced power factor in Ho3+ doped ZnO: A new material for TE application. AIP Conf. Proc. 1832, 110055 (2017)
Rubi, D., Fontcuberta, J., Calleja, A., Aragonès, Ll., Capdevila, X.G., Segarra, M.: Reversible ferromagnetic switching in ZnO:(Co, Mn) powders. Phys. Rev. B 75, 155322 (2007)
Rubi, D., Calleja, A., Arbiol, J., Capdevila, X.G., Segarra, M., Aragonès, Ll., Fontcuberta, J.: Structural and magnetic properties of ZnO:TM (TM: Co, Mn) nanopowders. Journal of Magnetism and Magnetic Materials 316, e211–e214 (2007)
Dakhel, A.A., El-Hilo, M.: Ferromagnetic nanocrystalline Gd-doped ZnO powder synthesized by coprecipitation. J. Appl. Phys. 107, 123905 (2010)
Kim, H.P., Yusoff, A.R., Lee, H.J., Lee, S.J., Kim, H.M., Seo, G.J., Youn, J.H., Jang, J.: Effect of ZnO:Cs2CO3 on the performance of organic photovoltaics. Nanoscale Res. Lett, 9, 323 (2014)
Divya, N.K., Pradyumnan, P.P.: Solid state synthesis of erbium doped ZnO with excellent photocatalytic activity and enhanced visible light emission. Mater. Sci. Semicond. Process. 41, 428–435 (2016)
Vaseem, M., Umar, A., Hahn, Y.B.: ZnO nanoparticles: Growth, properties, and applications. Metal Oxide Nanostruct. Appl. 5, 1–36 (2010)
Kumar, H., Rani, R.: Structural and optical characterization of ZnO nanoparticles synthesized by microemulsion route. Int. Lett. Chem. Phys. Astron. 19, 26–36 (2013)
Hernández, A., Maya, L., Sánchez-Mora, E., Sánchez, E.M.: Sol-gel synthesis, characterization and photo-catalytic activity of mixed oxide ZnO-Fe2 O 3. J. Sol-Gel Sci. Technol. 42, 71–78 (2007)
Das, J., Evans, I.R., Khushalani, D.: Zinc glycolate: a precursor to ZnO. Inorg. Chem. 48, 3508–3510 (2009)
Jagannatha Reddy, A., Kokila, M.K., Nagabhushana, H., Shivakumara, C., Chakradhar, R.P.S., Nagabhushana, B.M., Hari Krishna, R.: Luminescence studies and EPR investigation of solution combustion derived Eu doped ZnO. Spectrochimica Acta Part A: Molec. Biomolec. Spectros. 32, 305–312 (2014)
Anandan, S., Miyauchi, M.: Ce-doped ZnO (CexZn1−xO) becomes an efficient visible-light-sensitive photocatalyst by co-catalyst (Cu2+) grafting. Phys. Chem. Chem. Phys. 13, 14937–14945 (2011)
Zhang, F., Jin, Q., Chan, S.-W.: Ceria nanoparticles: size, size distribution, and shape. J. Appl. Phys. 95, 4319–4326 (2004)
Coey, J.M.D., Wongsaprom, K., Alaria, J., Venkatesan, M.: Charge-transfer ferromagnetism in oxide nanoparticles. J. Phys. D 41, 134012 (2008)
Phan, T. L., Zhang, Y.D., Yang, D.S., Nghia, N.X., Thanh, T.D., Yu, S.C.: Defect-induced ferromagnetism in ZnO nanoparticles prepared by mechanical milling. Appl. Phys. Lett. 102, 072408 (2013)
Phadnis, C., Inamdar, D.Y., Dubenko, I., Pathak, A., Ali, N., Mahamuni, S.: Ferromagnetic ZnO nanocrystals and Al-induced defects. J. Appl. Phys. 110, 114316 (2011)
Inamdar, D.Y., Lad, A.D., Pathak, A.K., Dubenko, I., Ali, N., Mahamuni, S.: Ferromagnetism in ZnO nanocrystals: doping and surface chemistry. J. Phys. Chem. C 114, 1451–1459 (2010)
Chen, W.B., Liu, X.C., Li, F., Chen, H.M., Zhou, R.W., Shi, E.W.: Influence of oxygen partial pressure on the microstructural and magnetic properties of Er-doped ZnO thin films. AIP Adv. 5, 067105 (2015)
Dietl, T., Ohno, H., Matsukura, F.: Hole-mediated ferromagnetism in tetrahedrally coordinated semiconductors. Phys. Rev. B 63, 195205 (2001)
Jayachandraiah, C., Sivakumar, K., Divya, A., Krishnaiah, G.: Erbium induced magnetic properties of Er/ZnO nanoparticles. AIP Conf. Proc. 1731, 050116 (2016)
Jadhav, S.S., Shirsath, S.E., Patange, S.M., Jadhav, K.M.: Effect of Zn substitution on magnetic properties of nanocrystalline cobalt ferrite. J. Appl. Phys. 108, 093920 (2010)
Topkaya, R., Baykal, A., Demir, A.: Yafet–Kittel-type magnetic order in Zn-substituted cobalt ferrite nanoparticles with uniaxial anisotropy. J. Nanopart. Res. 15, 1359 (2013)
Kung, C.Y., Young, S.L., Chen, H.Z., Kao, M.C., Horng, L., Shih, Y.T., Lin, C.C., Lin, T.T., Ou, C.J.: Influence of Y-doped induced defects on the optical and magnetic properties of ZnO nanorod arrays prepared by low-temperature hydrothermal process. Nanoscale Res. Lett. 7, 372–377 (2012)
Chikazumi, S.: Physics of Magnetism, p. 187. Wiley, N. Y. (1959)
Xavier, S., Thankachan, S., Jacob, B.P., Mohammed, E.M.: Effect of samarium substitution on the structural and magnetic properties of nanocrystalline cobalt ferrite. J. Nanosci. 2013, 524380 (2013)
Stoner, E.C., Wohlfarth, E.P.: A mechanism of magnetic hysteresis in heterogeneous alloys. Philos. Trans. R. Soc. London, Series A Math. Phys. Sci. 240, 599–642 (1948)
Vlasenko, L.S.: Point defects in ZnO: Electron paramagnetic reso- nance study. Physica B: Condensed Matter 404, 4774–4778 (2009)
Liu, W.K., Whitaker, K.M., Smith, A.L., Kittilstved, K.R., Robinson, B.H., Gamelin, D.R.: Room-temperature electron spin dynamics in free-standing ZnO quantum dots. Phys. Rev. Lett. 98, 186804 (2007)
Weil, J.A., Bolton, J.R.: Electron Paramagnetic Resonance, 2nd edn. Wiley, Hoboken (2007)
Yadav, N., Kumar, A., Rana, P.S., Rana, D.S., Arora, M., Pant, R.P.: Finite size effect on Sm3+ doped Mn0.5Zn0.5SmxFe2−x O 4 (0 x 0.5) ferrite nanoparticles. Ceram. Int. 41, 8623–8629 (2015)
Humánez, M.A., Montes-Vides, L., Almanza-Montero, O.: Sol-gel synthesis of zinc oxide nanoparticle at three different temperatures and its characterization via XRD, IR and EPR. DYNA 83, 224–228 (2016)
Acosta-Humánez, F., Cogollo Pitalúa, R., Almanza, O.: Electron paramagnetic resonance in Zn1−xCoxO. J. Magn. Magn. Mater. 329, 39–42 (2013)
Vlasenko, L.S.: Magnetic resonance studies of intrinsic defects in ZnO: oxygen vacancy. Appl. Magn. Reson. 39, 103–111 (2010)
Janotti, A., Van de Walle, C.G.: Native point defects in ZnO. Phys. Rev. B. 76, 165202 (2007)
Wang, J., Hou, S., Chen, H., Xiang, L.: Defects-induced room temperature ferromagnetism in ZnO Nanorods grown from 𝜖-Zn(OH)2. J. Phys. Chem. C118, 19469–19476 (2014)
Beltrán, J.J., Barrero, C.A., Punnoose, A.: Evidence of ferromagnetic signal enhancement in Fe and Co codoped ZnO nanoparticles by increasing superficial Co3+ content. J. Phys. Chem. C118, 13203–13217 (2014)
Orlinskii, S.B., Schmidt, J., Baranov, P.G., Lorrmann, V., Riedel, I., Rauh, D., Dyakonov, V.: Identification of shallow Al donors in Al-doped ZnO nanocrystals: EPR and ENDOR spectroscopy. Phys. Rev. B 77, 115334 (2008)
Yu, B., Zhu, C., Gan, F., Huang, Y.: Electron spin resonance properties of ZnO microcrystallites. Mater. Lett. 33, 247–250 (1998)
Jing, L., Xu, Z., Shang, J., Sun, X., Cai, W., Guo, H.: The preparation and characterization of ZnO ultrafine particles. Mater. Sci. Eng. A 332, 356–361 (2002)
Liu, X., Lin, F., Sun, L., Cheng, W., Ma, X., Shi, W.: Doping concentration dependence of room-temperature ferromagnetism for Ni-doped ZnO thin films prepared by pulsed-laser deposition. Appl. Phys. Lett. 88, 62508 (2006)
Elilarassi, R., Chandrasekaran, G.: Synthesis, structural and magnetic characterization of Ni-doped ZnO diluted magnetic semiconductor. Amer. J. Mater. Sci. 2, 46–50 (2012)
Alaria, J., Bouloudenine, M., Schmerber, G., Colis, S., Dinia, A.: Pure paramagnetic behavior in Mn-doped ZnO semiconductors. J. Appl. Phys. 99, 08M118 (2006)
Shahane, G.S., Kumar, A., Arora, M., Pant, R.P., Lal, K.: Synthesis and characterization of Ni–Zn ferrite nanoparticles. J. Magn. Magn. Mater. 322, 1015–1019 (2010)
Elilarassi, R.R., Chandrasekaran, G.: Structural, optical and electron paramagnetic resonance studies on Cu-doped ZnO nanoparticles synthesized using a novel auto-combustion method. Front. Mater. Sci. 7, 196–201 (2013)
Acknowledgements
This work has been performed in the materials science Lab, Physics Department, Faculty of Science, Beirut Arab University, Debbieh, in cooperation with the Faculty of Science, Alexandria University, Alexandria, Egypt.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Farhat, S., Rekaby, M. & Awad, R. Synthesis and Characterization of Er-Doped Nano ZnO Samples. J Supercond Nov Magn 31, 3051–3061 (2018). https://doi.org/10.1007/s10948-017-4548-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-017-4548-9