
Abstract
Pure and a series of rare earth element (RE) (Er, Sm, Gd, Pr and Y)-doped NiO nanoparticles (NPs) were fabricated by co-precipitation method in the presence of polyvinyl alcohol as a capping agent. X-ray diffraction (XRD) and transmission electron microscope (TEM) techniques were used to investigate the crystal structure and morphology of the prepared samples, respectively. The XRD patterns indicated that all the samples exhibited a single phase of face-centered cubic crystalline structure. TEM images displayed a spherical morphology of weakly agglomerated NPs. Furthermore, the chemical compositions were identified by proton-induced X-ray emission and Rutherford backscattering analysis. The metal oxide (Ni–O) functional group was confirmed by Fourier transform infrared spectroscopy with its observed band varying from 419.3 to 433.3 cm−1, depending on the RE doping element. The optical properties of the RE-doped NiO NPs were studied using UV–Vis absorption and photoluminescence (PL) spectroscopy. A small blueshift was obtained in the optical spectra of the RE-doped samples as compared to the undoped samples, implying an increase in the optical band gap. The study of the room temperature magnetic properties was done using the vibrating sample magnetometer, which revealed the coexistence of antiferromagnetic and weak ferromagnetic ordering in pure and RE3+-doped NiO NPs. The magnetization increased depending on the magnetic moments of RE dopant ions. Also, the origin of the anomalous ferromagnetism in the prepared samples may be mainly related to Ni vacancy defects, which were evinced from the results of PL.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Metal oxide nanoparticles, with particle size less than about 100 nm, have attracted scientists extensively because of their significant electronic, magnetic, thermal, catalytic and optical properties with respect to their bulk counterparts [1]. NiO is a p-type transparent semiconductor that has wide and direct band gap in the range of 3.6–4.0 eV [2]. With regard to its magnetic ordering, the bulk crystal of NiO exhibits antiferromagnetism (AFM) with a relatively high Neel temperature TN ∼ 523 K [3]. Because of the volume effect, the quantum size effect, the surface effect and the macroscopic quantum tunneling effect, NiO NPs are expected to possess many improved properties as compared to those of the bulk-sized NiO particles. In recent years, NiO NPs have many applications in catalysis [4], battery cathodes [5], gas sensors [6], electrochromic films [7], transparent conducting films [8] and dye-sensitized photocathodes [9]. In addition, the development of NiO NPs has focused on obtaining ferromagnetism (FM) for spintronic devices [10]. The magnetic properties of the nanostructures may be varied according to the synthesis method, antiparticle interaction, vacancies and nanoparticle size [11, 12]. Rinaldi-Montes et al. [13] reported that NiO, which is antiferromagnetic in its bulk form, exhibits ferromagnetic behavior at the nanoscale due to nickel vacancy defects, with its net magnetic moment increasing monotonically with decreasing particle size. Below a critical size of ~ 10 nm, as a consequence of the enhanced nickel vacancies, the Ni2+–O2−–Ni2+ superexchange interaction breaks down and the particles exhibit paramagnetic behavior at room temperature and superparamagnetic properties at lower temperature.
In order to obtain better optical and magnetic properties of NiO NPs for various applications, chemical doping has been adopted in the preparation techniques. The addition of RE dopants can finely tune these properties due to their partially filled inner (4fn) shell, surrounded by completely filled outer (5s2 and 5p6) orbitals [14]. Recently, Abdur Rahman et al. [15] have reported the doping effect of Ce3+ on the optical and magnetic properties of NiO NPs, prepared through chemical method. The Ce3+ doping altered the band emission of the pure NiO NPs due to nickel vacancies, oxygen vacancies and surface defects. Moreover, the magnetization values for Ce-doped NiO NPs were enhanced. Chauhan et al. [16] reported the synthesis and characterization of Nd3+-doped NiO NPs, prepared by sol–gel method. The results showed that the Nd3+-doped NiO samples show lower PL intensity than that of the pure NiO sample. This phenomenon may be due to the decrease in particle size. Similar results were obtained for La3+-doped NiO-sprayed thin films [17], where PL intensities decreased with decreasing crystallite size. This was contributed to the improvement in the sensitivity performance and photocatalytic applications of such doped films.
The purpose of this work is the synthesis of pure NiO and rare earth ions RE3+-doped NiO NPs by the co-precipitation process with the presence of polyvinyl alcohol (PVA), with a melting point between 180 and 240 °C [18], as capping agent. We have selected a few representative RE elements, such as Er, Gd, Sm, Pr and Y. The impact of RE3+ ions on the structural, optical and magnetic properties was investigated, through the different characterization facilities, to explore their feasibility for applications.
2 Experimental details
Different 2% RE-doped NiO NPs (Ni0.98RE0.02O) were prepared by co-precipitation method using salt chlorides, including NiCl2·6H2O, ErCl3·6H2O, SmCl3·6H2O, GdCl3·6H2O, PrCl3·xH2O and YCl3·6H2O, purchased from Sigma-Aldrich, as the starting materials. First, 1 M chloride solutions were prepared by weighing the powder salts according to stoichiometric calculations and dissolving them in distilled water. About 0.40 g/L polyvinyl alcohol PVA [CH2CH(OH)]n was added to each solution. Then, the solutions were titrated by 4 M NaOH until the pH reached 13. The obtained solutions were continuously stirred and heated at 60 °C for 2 h. In order to get rid of the residuals, the solutions were washed with distilled water until the pH reached about 7. Consequently, the obtained products were dried at 100 °C for 20 h, followed by grinding and calcination at 550 °C for 4 h. The six samples were prepared in parallel with the attempt of maintaining the same rate of titration, stirring and heating. Finally, the obtained powders were ball-milled, for 10 min at 400 rpm speed, with balls having a mass equals to five times the mass of the powder, in order to obtain a homogeneous grinding of the powder and to lower the extent of agglomeration.
The final powder was characterized and studied using several techniques. The structural properties such as the crystallinity, phase structure, lattice parameters, size and strain of the synthesized samples were studied with XRD patterns obtained from X-ray diffraction (XRD) technique. Bruker D8 Focus diffractometer, with CuKα radiation (λ = 1.54056 Å) in the range 20° ≤ 2θ ≤ 80° operating at 40 kV and 40 mA, was used where the powder was placed in a platelike shaped holder in a nearly continuous distribution of crystallites. Transmission electron microscopy (TEM) was done to the samples using JEM-100CX, operated at 80 kV for further structural and morphological characterization. The compositional proportions were checked simultaneously by proton-induced X-ray emission (PIXE) and Rutherford backscattering (RBS) using a 3 MeV proton beam provided by the 1.7 MV 5-SDH NEC horizontal, tandem accelerator. The sample, taking the dimensions of 1 × 1 × 0.20 cm3 thick pellets pressed with borax powder, was mounted on a rotating wheel and controlled by PC and stepping motor [16]. The functional groups were investigated by performing Fourier transform infrared spectroscopy (FTIR) with a Nicolet iS5 spectrometer on the samples mixed with KBr and pressed in disk shapes. The optical properties were studied using UV–Vis spectra and photoluminescence (PL) spectra recorded at room temperature using the ultraviolet near-infrared (NIR) V-670 spectrophotometer and FP-8300 spectrofluorometer, respectively. 0.015 g of the powder was dissolved in 20 ml of ethanol and placed in the cuvettes of the spectrometers. The data were then recorded in the range of 200–700 nm for the UV–Vis spectroscopy and 310-700 nm under excitation source of 280 nm, for the PL spectroscopy. Moreover, investigation of the magnetic properties was facilitated using the vibrating sample magnetometer (VSM) Lakeshore 7410 by recording the magnetization as a function of the applied magnetic field in the range from − 20 to 20 kG, at room temperature.
3 Results and discussion
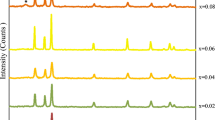
Figure 1 depicts the XRD patterns of the pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs, prepared under the same conditions via co-precipitation method. All samples exhibit characteristic diffraction peaks at angles 2θ ≈ 37.2°, 43.3°, 62.9°, 75.4° and 79.4°, corresponding to (111), (200), (220), (311) and (222) crystal planes, respectively. These peaks are indexed to the FCC structure with space group (Fm3m), JCPDS card No. 01-175-0269. In addition, the absence of any impurity peaks confirms the high purity of the prepared samples. Compared with the pure NiO sample, the highest diffraction peak (200) is noticed to shift slightly toward higher angles (inset in Fig. 1). This phenomenon could be associated with the shrinkage of the crystal lattice, which is strongly attributed to the ionic radii of the doping and host elements. The average lattice constant “a” of the pure and RE-doped NiO NPs with cubic lattice structure was calculated and is listed in Table 1. It is found that the average lattice constant of NiO NPs decreases when doped with RE3+ ions. As known, the ionic radii of RE3+ (Er3+ 0.0890 nm, Sm3+ 0.0958 nm, Gd3+ 0.0938 nm, Pr3+ 0.0990 nm and Y3+ 0.0900 nm) are greater than that of Ni2+ (Ni2+ 0.0690 nm) ions [19]. The decrease in lattice parameter with RE3+ doping ions can be attributed to several factors, including the concentration of dopant, defects (vacancies, interstitial, dislocation), external strains produced due to temperature and the large difference between the ionic radii of RE3+ and Ni2+ ions. Recently, Obeid et al. [20] have reported that for Zn1-xGdxO, the lattice contraction occurs up to a critical concentration (x = 0.03) of Gd3+ ions, due to the hydrostatic pressure created by the rare earth dopant located on the surface of the ZnO nanorods. However, the decrease in lattice parameter indicates that rare earth ions RE3+ are unable to substitute Ni sites completely, owing to the segregation of RE3+ ions to more energetically favorable grain boundary [14].

XRD patterns of pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs
From the XRD patterns, the intensity of the diffraction characteristic peaks was decreased upon doping with different RE3+ ions and the full width at half maximum (FWHM) became broader than that of the pure NiO NPs. The average crystallite size D, as related to the line broadening, can be calculated using the well-known Scherrer’s formula. The values of D are found to be around 9–15 nm, as listed in Table 1. The average crystallite size of RE3+-doped NiO samples is smaller than that of the pure NiO sample. This is due to the large difference in ionic radii between RE3+ and Ni2+ ions. Hence, the lattice disorder and strain induced in the NiO lattice triggers the change in growth kinetics and nucleation [21]. The X-ray density ρx, the dislocation density δ and specific surface area S were calculated and are listed in Table 1 using the following relations, respectively [22]:
where M is the molecular weight and N is Avogadro’s number. The density of the RE-doped NiO NPs is found to be higher than that of the pure NiO NPs. Generally, density depends on the unit cell volume of the crystal system. Here, the unit cell volume of the RE-doped NiO NPs is smaller than that of the pure NiO NPs, which in turn leads to higher density of the RE-doped NiO NPs. The dislocation densities increase with decreasing particle size as shown in Table 1; this is based on the fact that the amount of defects and vacancies in the crystal is defined as the length of dislocation lines per unit volume of the crystal [23]. In case of RE3+-doped NiO NPs, XRD shows lower peak intensities upon RE3+ doping, indicative of decrease in particle size, and therefore, the level of surface defects increases and crystal quality deteriorates [23, 24]. For Sm-doped NiO sample, the values of dislocation density and specific surface area are the highest.
In order to find the effect of strain on peak broadening, according to Williamson–Hall (W–H) method, the individual contribution to the line broadening β at the FWHM of Bragg reflection can be expressed as follows [25]:
where k is the shape factor and is usually taken as about 0.89 for spherical crystallites with cubic unit cell [26], βD is the peak broadening due to the contribution of crystallite size D and βɛ is the peak broadening due to the lattice strain ɛ. Equation (5) is Williamson–Hall (W–H) modified, which represents the uniform deformation model (UDM), where the strain is assumed to be uniform in all crystallographic directions. The strain and the average crystallite size can be predicted from the slope and the intercept of the plot between βcosθ against 4sinθ, respectively, as shown in Fig. 2. The positive slope value indicates the tensile strain. The scattered data in Fig. 2 show that the crystallite size and strain do not vary monotonically according to the W–H formula for different samples. This indicates that the broadening of the Bragg diffraction peaks with respect to the different Bragg reflections is anisotropic and is caused by an anisotropic microstrain. The obtained values from the UDM are summarized in Table 1. It is observed that the crystallite size calculated using Scherrer’s formula differs from that calculated using UDM by 3–11%. Furthermore, the strain values of the RE-doped NiO NPs are lower than that of the undoped NiO NPs, except for the Sm-doped NiO sample. However, the increase in the dislocation with the decrease in the lattice strain means that the X-ray diffraction peak broadening of the RE-doped samples is mainly due to the crystallite size reduction [27].

W–H plots of pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs
The particle size, shape and morphology of the pure and RE3+-doped NiO NPs are assured from transmission electron microscopy TEM. TEM images are displayed in Fig. 3a–f for pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs, respectively. It is clear that the samples consist of cluster islands, each consisting of several NPs that are in close contact and most of them is almost spherical and regular in shape. This indicates that the addition of PVA, the polymer capping agent, controls the size as well as the shape of nanoparticles and prevents their agglomeration [18]. Similar results were also observed by Tripathy et al. [28] for PVA capped silver NPs. Moreover, the average particle sizes obtained from TEM analysis, as listed in Table 1, are larger than those obtained from XRD analysis due to the weak agglomeration of the nanoparticles. The percentage difference varied between 64 and 2% for pure and the RE-doped NiO NPs, respectively. This is because the RE dopants of larger ionic radius tend to segregate to the surface more strongly and reduce the surface energy to a greater extent [29]. The particle size distribution histograms (shown in the insets in Fig. 3) show that the distribution is quite narrow within average size range of 12–19 nm (see Table 1) for RE-doped NiO NPs.

TEM images of a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs, capped with PVA. The inset graphs show the particle size distribution of the corresponding samples
Figure 4 depicts the PIXE spectra of the pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs. Ni and Y elements were identified by their characteristic K X-ray lines, while Er, Sm, Gd and Pr elements were identified by their characteristic L X-ray lines. Taking into account the errors and the uncertainties of the PIXE measurements, the stoichiometric real elemental contents are obtained and listed in Table 2. The uncertainties of the PIXE measurements are ± 2.88% for Ni, ± 1.66% for O, + 1.43% for Er, + 27.93% for Sm, + 26.75% for Gd, − 23.01% for Pr and + 9.45% for Y. The O content is determined indirectly by completing the 100 wt% of the obtained contents. In order to more accurately determine the O content, RBS analysis was done and the results are also listed in Table 2 with their corresponding spectra shown in Fig. 5 for all samples. The deviation between the two techniques is acceptable (less than 11%), and the obtained oxygen content shows that the doping was successfully done. However, the oxygen-rich stoichiometry of the prepared nanoparticles could possibly be attributed to the interstitial oxygen (Oi) defects in the NiO NPs [30]. This suggests that our prepared NiO NPs do not have interstitial nickel (Nii) defects, and therefore, Ni-deficient condition could consequently give rise to oxygen-rich stoichiometry of prepared nanoparticles [31].

PIXE spectra of a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs

RBS spectra of a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs
Figure 6 depicts the FTIR spectra of pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs measured in the wavenumber region of 400–4000 cm−1. The broad and sharp bands observed around 3441 and 1637 cm−1 are the characteristics of the hydroxyl (OH) group, which could be attributed to the O–H stretching and bending vibration of water molecules, respectively [32]. The two small peaks observed around 2365 cm−1 are due to O=C=O in air [33]. The strong absorption band in the range 409.3–433.4 cm−1 is the characteristic band of NiO NPs, corresponding to the stretching vibrational mode of Ni–O bonds, which confirms the formation of NiO NPs [15]. However, the NiO vibrational band is shifted toward higher value, depending on the RE3+ doping type. This shift is due to the variation of the ionic radii and, consequently, the lattice constant [34]. The removal of PVA is also confirmed by the absence of the two characteristic bands at 2920 and 2850 cm−1, corresponding to the asymmetric and symmetric CH2 stretching modes, respectively [35, 36]. Furthermore, these results demonstrate that the prepared samples have no traces of impurity, which is in agreement with the XRD results.

FTIR spectra of a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs
The optical absorption spectra of pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs, in the range 200–700 nm, are shown in Fig. 7. The inset in Fig. 7 shows the UV absorbance spectrum of the pure NiO NPs at 327 nm. A clear blueshift of the absorption edge with RE3+ doping was obtained at shorter wavelength in UV region at 317, 319, 313, 302 and 316 nm for Ni0.098Er0.02O, Ni0.098Sm0.02O, Ni0.098Gd0.02O, Ni0.098Pr0.02O and Ni0.098Y0.02O NPs, respectively. This blueshift reflects the increase in optical energy gap due to doping with RE3+ ions.

UV–Vis absorption spectra of pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs. The inset shows the absorption spectrum of pure NiO NPs
The absorbance can vary depending upon several factors like particle size, vacancy defects in the material prepared, etc. [37]. As shown in Fig. 7, the UV absorbance decreases upon doping with RE3+ ions. Similar results have been obtained for Ce3+-doped NiO NPs [15]. Regarding the particle size, Goh et al. [38] have shown that the UV absorbance of ZnO NPs decreases with decreasing their size in the range of 15–40 nm. It is well known that RE3+ dopant ions on the nanocrystal surfaces may inhibit growth and hence obtain smaller size [39], which in turn decreases the absorbance intensity. The particle size reduction has been confirmed by XRD and TEM analysis. The evolution of energy band gap is highly correlated with the calculated particle size values. It was reported that a small change in particle size distribution can contribute to a significant variation of the absorption edge, which will directly affect the optical band gap energy [40]. The optical band gap Eg can be evaluated using the well-known Tauc’s relation [22]. Figure 8a–f shows Tauc’s plots of (αhν)n versus the photon energy hν for the pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs, respectively. Here, α is the absorption coefficient, while n takes on the values of 2 and 1/2 in case of allowed direct and allowed indirect optical transitions, respectively. The extrapolation of the linear part of the curve in the high-absorption region gives the value of the band gap energy for direct \(E_{\text{gd}}\) and indirect \(E_{\text{gi}}\) band gaps on the x-axis. A more accurate position of the critical point of the energy band gap in the E-axis can be estimated from the second derivative curve of the absorbance versus E, as shown in the inset graphs in Fig. 8. The absorption onset \(E_{\text{g}}^{\text{onset}}\) (departure from zero in the second derivatives of the absorption spectra) is often used to define the optical gap. The estimated energy band gaps of the RE-doped NiO NPs exhibit higher values as compared to that of the pure NiO sample. The blueshift and the band gap increase can be explained by the quantum size confinement [41]. This leads to the increase in energy band gap whenever the particle size decreases. Furthermore, this blueshift in band gap can be ascribed to the Burstein–Moss effect [42], which originates from lifting of the Fermi level up to the conduction band by the generation of more carrier concentration, and hence, the optical absorption edge shifts to higher energy. So, increasing the carrier concentration by replacing Ni2+ with RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+) dopants in NiO host lattice will cause the Fermi level to move into the conduction band, and consequently, the optical band gap will be increased. The pure NiO has direct energy gap value of 3.11 eV, and the value is increased to 3.48 eV for Pr-doped NiO NPs. These results support our assumption that the RE3+ ions have successfully incorporated into the cubic structure of NiO and partially substituted the Ni cation sites.

Tauc’s plots of (αhν)n versus E of the a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs. The insets show the second derivative of optical absorbance versus E, which also allows to calculate the band gap energy. The arrows indicate the position of band gap energy
Moreover, the presence of both direct and indirect band gaps together in the same particles, with the indirect band gap energy \(E_{\text{gi}}\), lower than the direct band gap energy \(E_{\text{gd}}\), causes the better use as efficient catalysts for catalytic reactions under the full irradiation of solar spectrum. This can be provided by enough phonon-assisted indirect electron transition from the valence band (VB) to the conduction band (CB) [43].
Figure 9 depicts the room temperature PL emission spectra of pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs, excited using wavelength of 280 nm. Generally, PL emission peaks of NiO are located at 326, 365, 403, 471, 540 and 620 nm, respectively. Two UV emission peaks located at 326 and 365 nm correspond to near-band-edge emission (NBE). These peaks are due to the recombination of excitons corresponding to NBE transition of NiO NPs [44]. On the other hand, deep level emissions (DLE), in the visible region, can be related to various structural defects, such as Ni vacancies and oxygen interstitials present in the sample as a result of heat treatment and deviation from NiO stoichiometry [45]. The violet luminescence peak at 403 nm can be ascribed to possible energy transition of trapped electrons at Ni interstitial to the VB [46]. A strong broad blue emission centered at 471 nm is attributed to radiative recombination of electrons from the doubly ionized Ni vacancy (\({\text{V}}_{\text{Ni}}^{2 - }\)) to the holes in the VB [46]. The origin of the weak broad green emission, observed at 527 nm, is the most controversial. It is widely reported and attributed to the defects originated from NiO lattice such as interstitial oxygen trapping and Ni vacancies that can be produced due to charge transfer between Ni2+ and Ni3+ [47]. The very weak peak corresponding to orange–red emission at 620 nm is usually assigned to the O interstitials [48] due to the transition from CB to monovalent vacancies. This is consistent with the previous suggestions from RBS analysis.

PL spectra of pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs
Compared to the undoped NiO, the intensity of all the PL emissions decreases for the RE-doped NiO NPs. The suppression of the UV emissions, associated with RE doping, is due to the decrease in crystallite size [15, 16]. Therefore, small particles possessing high Ni vacancies on the surface (that is, a low electron population in the VB), when excited, will cause a reduction in PL intensity spectra as a result of the suppression of electron–hole recombination process [46]. The peaks due to DLE in the visible region are slightly redshifted and their intensities are also reduced with RE doping of NiO NPs. This quenching occurs maybe due to the photo-generated electrons trapped in the trap centers generated by the RE3+ ions [49, 50]. This causes the inhibition of more electrons (holes) to be excited and the enhancement of non-radiative recombination process. Moreover, trivalent RE3+ ions act as charge compensators to enter the surrounding of Ni2+ ions, which may lead to quenching of luminescence [51]. It is worth noticing here that the intensity of blue emission at 471 nm is more prominent than the green and orange–red emissions at 527 and 627 nm, respectively. Therefore, the dominant point defects in the prepared NiO NPs are Ni vacancies.
Figure 10 depicts the magnetic hysteresis (M–H) loops of pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs at room temperature. It is known that the bulk crystal of NiO is antiferromagnetic (AFM) with TN of ~ 523 K [3]. However, some reports suggested that NiO should become either weak ferromagnetic (FM) or superparamagnetic (SPM) after size reduction [52,53,54]. From the as-measured (M–H) curves, shown in Fig. 10, we can observe that all the samples display a linear antiferromagnetic (AFM) contribution in addition to a weak ferromagnetic (FM) signal at low fields, and no saturation of magnetization appears even at the maximum applied field of 20 kG. Even though the M–H loop exhibits no saturation, the presence of a small hysteresis loop in all the samples represents weak ferromagnetic nature in the NiO NPs (insets in Fig. 10). The mechanism responsible for the origin of ferromagnetism at room temperature in AFM NiO is still controversial. Up to date, the most commonly accepted mechanism is the vacancy-induced FM for AFM NiO NPs [55]. Nevertheless, the type of vacancy is still a debatable issue, as it is whether the FM is due to cation or anion vacancies effects. For instance, Madhu et al. [56] reported that for NiO NPs, the FM interaction was correlated with the formation of both uncompensated surface O2− and Ni2+ vacancies in NiO. On the other hand, Sugiyama et al. [57] demonstrated that FM properties in AF NiO NPs are due to the local non-stoichiometry of the dislocation cores with Ni deficiency, i.e., Ni vacancy.

Typical M–H hysteresis loops of a pure, b Er-doped, c Sm-doped, d Gd-doped, e Pr-doped and f Y-doped NiO NPs. The insets show enlarged detail of the hysteresis loop around the origin
In our study, the origin of this ferromagnetism of pure and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs may be mainly induced by vacancy defects on the surface of nanoparticles. According to the above PL analysis, the possible vacancy defects were Ni-related vacancies. These Ni vacancies may play important roles in generating FM, e.g., by polarizing the surrounding ligand atoms of the vacancy to form a localized magnetic moment. The FM interaction between two localized moments induces a collective ferromagnetic behavior depending on the concentration and the charge state (negative or neutral) of vacancy defects. The above results are supported by theoretical predictions of FM in NiO. Zhang et al. [58] reported that the FM of NiO originated from singly and/or doubly negative Ni vacancies under oxygen-rich stoichiometry. Mandal et al. [59] found that anomalous FM behavior in NiO NPs originates from Ni vacancies on the surface of nanograins, and the observed ferromagnetic enhancement in NiO NPs is not a true nanoscale effect, but rather is mainly determined by the surface vacancy defects. As for the origin of FM in NiO NPs, it was also suspected to result from the presence of metallic Ni clusters or impurity FM phases in NiO lattice [60]. Whatever, the above XRD results indicate that there is no trace of metallic Ni or any other ferromagnetic impurities such as Gd cluster or secondary phase of Gd2O3 in our sample within the detection limit of XRD. It is surprising to note that although Y3+ ion is non-magnetic (vacant d orbital), the Y-doped NiO NPs has also weak ferromagnetic order. Lan et al. [61] reported that the origin of ferromagnetism for Y3+-doped ZnO thin film is due to the Y3+ ion that favors the formation of Zn vacancies VZn. Therefore, we conclude that the Ni-related vacancy defect is mainly responsible for the FM in pure and RE3+-doped NiO NPs and not because of metal ions segregation.
For comparison, from the (M–H) curves of pure and RE3+-doped NiO NPs, the magnetic parameters such as saturation magnetization Ms, remanent magnetization Mr and coercivity Hc were calculated and are listed in Table 3. The saturation magnetization was determined using the law of approach to saturation (LAS) [62]. Figure 11 shows the plot of M versus 1/H2 for NiO and RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs. The plotted data are fitted by a straight line whose y-intercept determines the values of magnetization. The results indicate that the magnetization values can be enhanced upon doping with RE3+ ions. This is due to their dependence on the magnetic moments of RE3+ dopant ions. The samples doped with Gd3+ (8µB) and Er3+ (9.59 µB) [63] have higher magnetization values as compared to Pr3+ (3.58 µB)-, Sm3+ (0.85µB)- and Y3+ (0.0µB)-doped [63] NiO NPs. Moreover, these results are consistent with the PL spectra, where the peak intensity of the blue emission at 417 nm is higher for Er3+ and Gd3+ than for Pr3+-, Sm3+- and Y3+-doped NiO NPs. This implies that the reduction in the Ni vacancies for the Pr3+-, Sm3+- and Y3+-doped NiO NPs is more prominent than that for the Er3+- and Gd3+-doped NiO NPs, thereby decreasing their magnetic moments more. Thus, the RE-doped NiO NPs are tunable by RE3+ ions and can be employed in various spintronic device applications [10].

Variation of M versus 1/H2 curve of pure and rare earth ions RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs
The values of Mr and Hc for pure and RE3+-doped NiO NPs, estimated from the hysteresis curves, are listed in Table 3. The values of Mr are higher for the RE3+ (Er3+, Sm3+, Gd3+, Pr3+ and Y3+)-doped NiO NPs than that of the undoped NiO NPs, while Hc values are lower for Er3+ and Gd3+ ions and higher for Sm3+-, Pr3+- and Y3+-doped NiO NPs as compared to undoped NiO.
The lower values of Hc are due to the effect of particle size on the magnetic coercivity. Particle sizes that are smaller than a critical value Dc are known as “single-domain (SD) particles” [64]. For NiO NPs, the single-domain critical size is 21.2 nm as calculated by the magnetic domain theory [65], which is larger than the particle size of all our doped NiO samples. In the single-domain region, as the particle size decreases, the coercivity decreases because of the thermal fluctuating effects [66]. The decrease in Hc for Er- and Gd-doped NiO NPs, with decreasing particle size, has been reported for single-domain nanoparticles.
The higher values of Hc for Sm-, Pr- and Y-doped NiO NPs are mainly related to the increase in magnetocrystalline anisotropy K. This is because in single-domain particles or particles with low magnetization, the crystal structure dominates the magnetic energy [67]. In the case of non-interacting and randomly oriented spherical particles with cubic anisotropy, Hc obeys the relation [68]:
The values of K for pure and doped NiO NPs are listed in Table 3. The Y-doped NiO sample has the highest K value. The squareness ratio is calculated from the following equation [69]:
The squareness ratio is an indication of uniaxial anisotropy contribution in RE-doped NiO NPs generated by the internal strains [70]. High uniaxial magnetocrystalline anisotropy is a key prerequisite and a necessary condition for high coercivity [71]. From Table 3, the squareness values are less than 1, which is an indication of single-domain isolated ferromagnetism [72].
4 Conclusion
We have successfully synthesized NiO and RE (Er, Sm, Gd, Pr and, Y)-doped NiO via the co-precipitation method using PVA as a capping agent. The structural, morphological, optical and magnetic properties have been investigated for the prepared samples. The XRD patterns revealed that the synthesized nanoparticles exhibit a single-phase cubic structure. Further, the crystalline size evaluated from W–H plots and Scherer’s formula was slightly different. The crystallite size was decreased, and therefore, structure defects and dislocations were created due to the higher ionic radii of RE dopants. The spherical morphologies of the samples were observed by TEM analysis and affirmed that the particle size decreases by RE doping. PIXE and RBS analysis indicated that the values of the stoichiometric ratio of all compositional elements were close to the real one. Interestingly, oxygen-rich stoichiometry of nanoparticles was detected via RBS spectrum. FTIR supported the XRD data and confirmed the formation of NiO and RE-doped NiO NPs with high purity. A blueshift of the absorption peaks was observed upon RE doping, indicating an increase in the optical band gap. The cause of weak FM of pure and RE-doped NiO NPs can be principally attributed to the Ni vacancies, which is confirmed from PL spectra. The observation of FM in RE-doped NiO may have potential application in the field of spintronics.
References
S.M. Chavali, M.P. Nikolova, SN Appl. Sci. 1, 607 (2019)
H. Ramachandran, M.M. Jahanara, N.M. Nairab, P. Swaminathan, RSC Adv. 10, 3951 (2020)
A.C. Gandhi, S.Y. Wu, AIP Adv. 10, 015211 (2020)
M.S. Alnarabiji, O. Tantawi, A. Ramli, N.A.M. Zabidi, O.B. Ghanem, B. Abdullahad, Renew. Sustain. Energy Rev. 114, 109326 (2019)
S. Burrola, M. Horii, M.J. Gonzalez-Guerrero, J.C. Bachman, F.A. Gomez, Electrophoresis 41, 131 (2020)
N. Kaur, D. Zappa, E. Comini, Electron. Mater. Lett. 15, 743 (2019)
M. Da Rocha, A. Rougier, Appl. Phys. A 122, 370 (2016)
C. Guillén, J. Herrero, Mater. Res. Express 7, 1 (2019)
J. Massin, M. Bräutigam, S. Bold, M. Wächtler, M. Pavone, A.B. Munoz-Garcia, J. Phys. Chem. C 123(28), 17176 (2019)
P. Ravikumar, B. Kisan, A. Perumal, AIP Adv. 5, 087116 (2015)
A.C. Gandhi, T.S. Chan, J. Pant, S.Y. Wu, Nanoscale Res. Lett. 12, 207 (2017)
E. Erdem, K. Kiraz, M. Somer, R.A. Eichel, J. Eur. Ceram. Soc. 30, 289 (2010)
N. Rinaldi-Montes, P. Gorria, D. Martinez-Blanco, A.B. Fuertes, L.F. Barquin, J.R. Fernandez, I. de Pedro, M.L. Fdez-Gubieda, J. Alonso, L. Olivi, G. Aquilanti, J.A. Blanco, Nanoscale 6, 457 (2014)
A.S. Varpe, M.D. Deshpande, Pramana 89, 15 (2017)
M. Abdur Rahman, R. Radhakrishnan, SN Appl. Sci. 1, 221 (2019)
J. Chauhan, U. Chaubey, IOSRJEN 8, 88 (2018)
C. Mrabet, M. Ben Amor, A. Boukhachem, M. Amlouk, T. Manoubi, Ceram. Int. 42, 5963 (2016)
J. Junaidi, M. Yunus, H. Harsojo, E. Suharyadi, K. Triyana, Int. J. Adv. Sci. Eng. Inf. Technol. 6, 365 (2016)
J.A. Dean, Lange’s Handbook of Chemistry, 15th edn. (McGraw-Hill, New York, 1999)
M.M. Obeid, H.R. Jappor, K. Al-Marzoki, I.A. Al-Hydary, S.J. Edrees, M.M. Shukur, RSC Adv. 9, 33207 (2019)
P. Kumar, V. Sharma, A. Sarwa, A. Kumar, Surbhi, R. Goyal, K. Sachdev, S. Annapoorni, K. Asokan, D. Kanjilal, RSC Adv. 6, 89242 (2016)
J. Al Boukhari, L. Zeidan, A. Khalaf, R. Awad, Chem. Phys. 516, 116 (2019)
M. Gomathi, P.V. Rajkumar, A. Prakasam, Results Phys. 10, 858 (2018)
M.S. Geetha, H. Nagabhushana, H.N. Shivananjaiah, J. Sci. Adv. Mater. Dev. 1, 301 (2016)
P. Bindu, S. Thomas, J. Theor. Appl. Phys. 8, 123 (2014)
A. Monshi, M.R. Foroughi, M.R. Monshi, World J. Nano Sci. Eng. 2, 154 (2012)
P.K. Giri, S. Bhattacharyya, D.K. Singh, R. Kesavamoorthy, B.K. Panigrahi, K.G.M. Nair, J. Appl. Phys. 102, 093515 (2007)
S. Tripathy, Nanoscale Res. Lett. 3, 164 (2008)
MdM Hasan, P.P. Dholabhai, R.H.R. Castro, B.P. Uberuaga, Surf. Sci. 649, 138 (2016)
A. Janotti, C.G.V. de Walle, Rep. Prog. Phys. 72, 126501 (2009)
A. Sahai, N. Goswami, Ceram. Int. 40, 14569 (2014)
M. El-Kemary, N. Nagy, I. El-Mehasseb, Mater. Sci. Semicond. Process. 16, 1747 (2013)
T. Anantha Kumar, S. Malathi, C.V. Mythili, M. Jeyachandran, IJCTR 11, 48 (2018)
M.N. Akhtar, T. Hussain, M.A. Khan, M. Ahmad, Results Phys. 10, 784 (2018)
V. Singh, V. Srinivas, S. Ram, Philos. Mag. 90, 1401 (2010)
A.B. Gemta, B. Bekele, A.R.C. Reddy, Int. J. Nanotechnol. Mater. Sci. 5, 44 (2018)
T.N. Soitah, Y. Chunhui, S. Liang, Sci. Adv. Mater. 2, 534 (2010)
E.G. Goh, X. Xu, P.G. McCormick, Scripta Mater. 78–79, 49–52 (2014)
A. Layek, S. Banerjee, B. Manna, A. Chowdhury, RSC Adv. 6, 35892 (2016)
S. El Whibi, L. Derbali, P. Tristant, C. Jaoul, M. Colas, R. Mayet, J. Cornette, H. Ezzaouia, J. Mater. Sci. Mater. Electron. 30, 2351 (2019)
S.M. Meybodi, S.A. Hosseini, M. Rezaee, S.K. Sadrnezhaad, D. Mohammadyani, Ultrason. Sonochem. 19, 841 (2012)
K. Varunkumar, R. Hussain, G. Hegde, A.S. Ethiraj, Mater. Sci. Semicond. Process. 66, 149 (2017)
N. Saha, A. Sarkar, A.B. Ghosh, A.K. Dutta, G.R. Bhadu, P. Paul, B. Adhikary, RSC Adv. 5, 88848 (2015)
B. Kisan, P.C. Shyni, S. Layek, H.C. Verma, D. Hesp, V. Dhanak, S. Krishnamurthy, A. Perumal, IEEE Trans. Magn. 50, 1 (2014)
A. Diallo, K. Kaviyarasu, S. Ndiaye, B.M. Mothudi, A. Ishaq, V. Rajendran, M. Maaza, Green Chem. Lett. Rev. 11, 166 (2018)
A.C. Gandhi, S.Y. Wu, Nanomaterials (Basel) 7, 231 (2017)
B. Karthikeyan, T. Pandiyarajan, S. Hariharan, M.S. Ollakkan, Cryst. Eng. Comm. 18, 601 (2016)
X. Liu, X. Wu, H. Cao, R.P.H. Chang, J. Appl. Phys. 95, 3141 (2004)
M.V. Limaye, S.B. Singh, R. Das, P. Poddar, S.K. Kulkarni, J. Solid State Chem. 184, 391 (2011)
S.-Y. Kuo, W.-C. Chen, F.-I. Lai, C.-P. Cheng, H.-C. Kuo, S.-C. Wang, W.-F. Hsieh, J. Cryst. Growth 287, 78 (2006)
D.F. Crabtree, Phys. Status Solidi (a) 38, 217 (1976)
W.J. Duan, S.H. Lu, Z.L. Wu, Y.S. Wang, J. Phys. Chem. C 116, 26043 (2012)
Y. Shimomura, I. Tsubokawa, M. Kojima, J. Phys. Soc. Jpn. 9, 521 (1954)
S.A. Makhlouf, F.T. Parker, F.E. Spada, A.E. Berkowitz, J. Appl. Phys. 81, 5561 (1997)
Z.-Y. Chen, Y. Chen, Q.K. Zhang, X.Q. Tang, D.D. Wang, Z.Q. Chen, P. Mascher, S.J. Wang, ECS J. Solid State Sci. Technol. 6, P798 (2017)
G. Madhu, K. Maniammal, V. Biju, Phys. Chem. Chem. Phys. 18, 12135 (2016)
I. Sugiyama, N. Shibata, Z. Wang, S. Kobayashi, T. Yamamoto, Y. Ikuhara, Nat. Nanotechnol. 8, 266 (2013)
W.-B. Zhang, N. Yu, W.-Y. Yu, B.-Y. Tang, Eur. Phys. J. B 64, 153 (2008)
S. Mandal, S. Banerjee, K.S.R. Menon, Phys. Rev. B 80, 214420 (2009)
J.T. Richardson, D.I. Yiagas, B. Turk, K. Forster, M.V. Twigg, J. Appl. Phys. 70, 6977 (1991)
Z.H. Lan, X.J. Miao, in Applied Mechanics and Materials (Trans Tech Publ, 2014), pp. 70–73
A. Awadallah, S.H. Mahmood, Y. Maswadeh, I. Bsoul, M. Awawdeh, Q.I. Mohaidat, H. Juwhari, Mater. Res. Bull. 74, 192 (2016)
N.A. Spaldin, Magnetic Materials Fundamentals and Applications, 2nd edn. (Cambridge University Press, Cambridge, 2011)
M. George, A. Mary John, S.S. Nair, P.A. Joy, M.R. Anantharaman, J. Magn. Magn. Mater. 302, 190 (2006)
K.O. Moura, R.J.S. Lima, A.A. Coelho, E.A. Souza-Junior, J.G.S. Duque, C.T. Meneses, Nanoscale 6, 352 (2013)
S. Chakraverty, M. Bandyopadhyay, J. Phys. Condens. Matter 19, 216201 (2007)
R.M.H. New, R.F.W. Pease, R.L. White, IEEE Trans. Magn. 31, 3805 (1995)
S. Laureti, G. Varvaro, A.M. Testa, D. Fiorani, E. Agostinelli, G. Piccaluga, A. Musinu, A. Ardu, D. Peddis, Nanotechnology 21, 315701 (2010)
R. Tholkappiyan, K. Vishista, Appl. Surf. Sci. 351, 1016 (2015)
S. Ammar, A. Helfen, N. Jouini, F. Fiévet, I. Rosenman, F. Villain, P. Molinié, M. Danot, J. Mater. Chem. 11, 186 (2001)
S. Liu, Chinese Phys. B 28, 017501 (2019)
N.S. Kumar, K.V. Kumar, Soft Nanosci. Lett. 6, 37 (2016)
Acknowledgements
This work was performed in the materials science laboratory, Physics Department, Faculty of Science, Beirut Arab University, in collaboration with the Faculty of Science, Alexandria University, Alexandria, Egypt.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Al Boukhari, J., Khalaf, A., Sayed Hassan, R. et al. Structural, optical and magnetic properties of pure and rare earth-doped NiO nanoparticles. Appl. Phys. A 126, 323 (2020). https://doi.org/10.1007/s00339-020-03508-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-020-03508-3