
Abstract
Iron-doped SnO2 diluted magnetic semiconducting powders (Sn1−x Fe x O2, x=0.00, 0.03, 0.05, 0.07, 0.10, and 0.15) were synthesized by a simple solid state reaction followed by vacuum annealing and studied the effect of Fe dopant concentrations on structural, optical, and magnetic properties of the synthesized samples. From the X-ray diffraction, it was confirmed that the samples prepared at lower dopant concentrations were tetragonal in structure whereas the samples prepared at higher dopant concentration exhibited orthorhombic SnO and Fe2O3 phases along with tetragonal SnO2 structure. FT-IR spectrum has been used to confirm the formation of Sn–O bond. The optical band gap of the Sn1−x Fe x O2 powders was increased from 3.6 eV to 3.7 eV with increase of dopant concentration. Raman spectroscopy measurement revealed that the broadening of the most intense Raman peak observed at 630 cm−1 with Fe doping, conforming that the Fe ions are substituted at the Sn sites in the SnO2 lattice. Vibrating sample magnetometer measurements confirmed that the Sn1−x Fe x O2 powders were ferromagnetic at room temperature.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Since the discovery of room temperature ferromagnetism in Mn doped ZnO and GaN by Dietl et al. [1], research for the new kind of materials exhibiting ferromagnetism at/above room temperature are finding intense interest. A number of different semiconducting host materials have been investigated. Among which more attention was paid on (Ga, Mn)As [2] and (In, Mn)As [3] to make Curie temperature (T c) above room temperature, but reported the highest Curie temperature (T c) of 170 K for (Ga, Mn)As [4] and 35 K for (In, Mn)As [5], which is not suitable for practical applications. Hence, intense interest is being put in developing a new type of DMS materials, which exhibit ferromagnetism at/above room temperature. Oxide-based diluted magnetic semiconductors (DMS) are ones which can be best suited for the applications such spintronics, nanoelectronics, magnetoelectronics, and microwave devices [6, 7]. The advantages of oxide-based DMS are the wide band gap suits for applications of short wavelength light, capability to be grown on various different of substrates including plastic even at low temperatures, environment safety and durability, and low cost. Moreover, oxygen is expected to produce strong p−d exchange coupling between band carriers and localized spins [8]. Wide band gap oxides materials, such as TiO2, SnO2, In2O3, and ZnO doped with different transition metals (TM) opened the doors for the above said electronic devices because of their high optical transparency, electrical conductivity along with ferromagnetism at room temperature [9–16]. The ferromagnetism had been observed in oxide-based DMS systems in powder, nanoparticles, and thin film forms [17–19]. Among the different oxide materials, SnO2 is the material, which finds many optoelectronic applications such as SnO2 has been studied extensively because of its high optical transparency and high electrical conductivity with wide band gap (3.5 eV). Fitzgerald et al. [20] have reported the room temperature ferromagnetism in Sn1−x M x O2 (M = Mn, Fe, Co, x=0.05) ceramics and found that Sn1−x Co x O2 were paramagnetic whereas Sn1−x Mo x O2, Sn1−x Fe x O2 were ferromagnetic in nature. Sharma et al. [21] prepared Ni and Fe codoped SnO2 nanoparticles by the simple wet chemical method and reported that the undoped SnO2 as diamagnetic, whereas Ni and Fe doped SnO2 nanoparticles were ferromagnetic in nature. The present study is an attempt to impart room temperature ferromagnetism in Fe doped SnO2 and to study the origin of ferromagnetism. Different physical [22–24] and chemical methods [25, 26] have been applied for the synthesis of Fe doped SnO2 powders. However, very few works have explored the solid state reaction method. A simple solid state reaction method is used here to prepare Fe doped SnO2 powders and studied the influence of doping levels of Fe on structural, optical, and magnetic properties of (Sn1−x Fe x O2, x=0.00, 0.03, 0.05, 0.07, 0.10, and 0.15) powders.
2 Experimental
Sn1−x Fe x O2 powder samples (x=0.03, 0.05, 0.07, 0.10, and 0.15) were prepared by a solid state reaction followed by vacuum annealing. Commercially available SnO2 and Fe2O3 (M/S Sigma-Aldrich 99.99 % pure) were accurately weighed in required proportions and were mixed and ground thoroughly using an agate mortar and pestle to convert to very fine powders. The grinding of the mixtures was carried out for 16 hours for all the powder samples. The ground powder samples were loaded into a small one end closed quartz tube of diameter 10 mm and length of 10 cm, which was enclosed in a bigger quartz tube of diameter of 2.5 cm and length of 75 cm with provision to allow unwanted vapors to escape from the reaction chamber and evacuated at 2×10−3 mbar using a rotary pump was used for the synthesis of the present samples. The complete set up was placed in horizontal tubular microprocessor controlled furnace and fired for several hours at different temperatures. The firing temperature and firing periods were optimized at 900 °C and 10 hours. X-ray diffraction (X-ray diffractometer, D8 Advance, BRUKER) was used to establish structural aspects. Energy dispersive analysis spectroscopy (EDS) (OXFORD instrument inca penta FET X3) was used to carry out elemental analysis. The diffused reflectance spectra were recorded on UV-Vis-NIR Spectrophotometer (JASCO V-670). Fourier Transform Infrared (FT-IR) Spectroscopic analysis was carried using FT-IR Spectrophotometer (SHIMADZU). Magnetic measurements were carried out using Vibrating sample magnetometer (Lake Shore-7410, IIT Madras).
3 Results and discussion
3.1 Structural properties
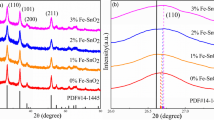
Figure 1(a) shows the X-ray diffraction patterns of the Sn1−x Fe x O2 at different Fe doping levels (x=0.03, 0.05, 0.07, 0.10, and 0.15). All the diffraction peaks having higher and lower intensities were taken into the considerations. The diffraction peaks were found at diffraction angles of 26.62∘ (1 1 0), 33.89∘ (1 0 1), 37.98∘ (2 0 0), 39.09∘ (1 1 1), 42.65∘ (2 1 0), 51.80∘ (2 1 1), 54.79∘ (2 2 0), 57.85∘ (0 0 2), 61.92∘ (3 1 0), 64.75∘ (1 1 2), 65.98∘ (3 0 1), 71.39∘ (2 0 2), and 78.73∘ (3 2 1) were exactly coincided with tetragonal structure of SnO2 [JCPDS No. 411445]. Among the above orientations, (1 1 0) is the predominant orientation. Same orientations were observed when the dopant concentration increased from 0.00 to 0.07. This clearly indicated that all Sn1−x Fe x O2 powder samples were polycrystalline in nature without having any secondary phase at lower Fe doping concentrations (x≤0.07). At a dopant concentration of 0.10, instead of single phase, other phases belonging to SnO orthorhombic were also identified. At higher Fe dopant concentration x=0.15, in addition to orthorhombic SnO phases, phases belonging to Fe2O3 were also observed. Further, as the Fe dopant concentration increased above 0.07, the intensity of the X-ray diffraction peaks decreased and FWHM increased, which show the degradation of crystallinity due to the additional impurities in the samples.

(a) X-ray diffraction patterns of Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, 0.07, 0.10, and 0.15. (b) Rietveld refinement patterns of Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, 0.07. Dots represent the observed intensities, and the solid line is calculated ones. A difference (obs.-cal.) plot is shown beneath. Vertical bars are the reflection position marks. (c) Rietveld refinement patterns of Sn1−x Fe x O2 powder samples at x=0.10 and 0.15. Dots represent the observed intensities, and the solid line is calculated ones. A difference (obs.-cal.) plot is shown beneath. Vertical bars are the reflection position marks
In order to further confirm that no Fe or Fe2O3 phases were present at lower Fe doping concentrations, Rietveld refinement analysis was also carried out at higher and lower Fe doping concentrations. From Fig. 1(b), it is clear that no other phases or impurities were observed when the Fe doping concentration was increased from x=0.00 to x=0.07, whereas impurity phases were observed at higher Fe doping concentrations as shown in Fig. 1(c). It clearly reflects that the miscibility of dopant impurity is limited in SnO2 lattice. It is necessary to note that the small limiting solubility of various metal oxides is typical for SnO2 [27]. Similar results were observed in Fe doped SnO2 by Mathew et al. [28]. The diffraction peaks angle (2θ) shifted slightly toward the lower angle with the Fe doping concentration. It may due to smaller ionic radius of Fe+3 (0.63 Å) than that of ionic radius of Sn+4 (0.71 Å).
The crystallite size (G) was calculated by using the Debye–Scherrer formula [29],
where k is a constant, λ is the diffraction wavelength of CuK α (λ=1.5406 Å), β is the full width at half maximum (FWHM), and θ is the diffracted angle, respectively. The grain size of the powder samples increased from 45 nm–92 nm with an increase of Fe doping concentration from 0.03 to 0.07. A decrease in lattice constant was observed from 4.7451 Å to 4.7358 Å when Fe doping concentration increased from 0.03 to 0.07. Further, the lattice volume decreased from 71.74 Å3 to 71.39 Å3 as the Fe doping concentration increased from x=0.03 to x=0.15. A summary of Fe doping concentration, grain size, lattice parameters “a” and “c” and lattice volumes is given in Table 1.
In order to know the elemental compositions of tin, oxygen and iron, EDS spectra were recorded for all the Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, 0.07, 0.10, and 0.15. Figure 2 show the EDS spectrum of Fe doped SnO2 powder sample at x=0.07. From the EDS spectra, it was found that the impurity was present in all the samples, which clearly represents the existence of Fe ions in doped sample and confirms the doping of Fe in SnO2 host material. The elemental compositions of Fe, Sn, and O were found to be as 1.06 %, 16.33 %, and 82.61 %, respectively.

EDS spectrum of Sn1−x Fe x O2 powder sample at x=0.07
3.2 Optical properties
Figure 3(a) shows the diffused reflectance spectra of the Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, 0.07. In addition to reflectance, absorbance spectra was also plotted (not shown here) for calculating the band gap of the Fe doped SnO2. The absorption coefficient was calculated using Kubelka–Munk equation [30],
where α is the absorption coefficient, R is reflectance of the samples. As secondary phases belonging to SnO and Fe2O3 were observed at Fe dopant concentration of x=0.10 and x=0.15, the diffused reflectance spectra and band gap values were not taken into considerations. The optical band gap (E g) of the powders were determined by Tauc’s plots using equation [31],
where n depend on the kind of optical transition that prevails. Here, n=1/2, as SnO2 is directly allowed n-type degenerate semiconductor. The optical band gap is obtained by plotting (αhυ)2 versus the photon energy (hυ) and by extrapolating of the linear region of the plots to zero absorption (α=0). The optical band gap E g is obtained by plotting (αhυ)2 versus the photon energy (hυ) and by extrapolating the linear region of the plots to zero absorption (α=0). The optical band gap of the powder samples increased from 3.6 eV to 3.7 eV when the Fe doping concentration increased from x=0.03 to x=0.07. At higher doping concentrations, SnO and Fe2O3 phases were present, hence the optical band gap at higher doping concentrations is not considered here. A similar increase in the optical band gap was also observed in Mn doped SnO2 nanoparticles and the observed band gap is lesser when compared with Mn doped SnO2 nanoparticles [32].

(a) Diffused reflectance spectra of Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, and 0.07. (b) Plots of (αhυ)2 versus hυ of Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, and 0.07
3.3 FT-IR studies
Fourier transform infrared spectra were recorded to pure Fe2O3 and Sn1−x Fe x O2 powder samples at x=0.00, 0.07, and 0.15 as shown in Fig. 4. The bands observed in the wavenumber range of 430 cm−1 to 620 cm−1 related to the vibration of antisymmetric O–Sn–O bridge functional groups of SnO2. The change in peak position, shape, and size indicate that incorporation of Fe took place in SnO2 host material.

FT-IR spectra of Fe2O3 and Sn1−x Fe x O2 powder sample at x=0.00, 0.07, and 0.15
3.4 Raman analysis
Raman scattering is one of the most powerful tool to study the crystallinity, phase composition, and defect structures associated with the material. Figure 5 shows the Raman spectra of Sn1−x Fe x O2 powders at x=0.00, 0.03, 0.05, and 0.07. Raman spectra of Sn1−x Fe x O2 powders at higher Fe doping concentrations are not shown here as secondary phases were observed at higher doping levels. The representation of the normal vibration modes at the center of the Brillouin zone were explained using Hong et al. [33]. The pure SnO2 spectrum shows the classic cassirite SnO2 vibration at 476, 630, and 776 cm−1. The peaks at 621 cm−1 can be assigned to the \(\mathrm{A}_{\rm 1g}\) mode, which is shifted to 608 cm−1, 606 cm−1, and 620 cm−1 for Sn1−x Fe x O2 powders at x=0.00, 0.03, 0.05, and 0.07, respectively. The Raman bands at 466 cm−1 and 762 cm−1 are assigned to be the vibration modes of E g and B 2g, respectively.

Raman spectra of Sn1−x Fe x O2 powder sample at x=0.00, 0.03, 0.05, and 0.07
3.5 Magnetic properties
Figure 6(a)–(c) shows the M–H curves for Sn1−x Fe x O2 powder sample with different Fe concentrations. Magnetization measurements were carried out for all the samples including pure SnO2. The intrinsic SnO2 exhibited diamagnetism. By introducing the Fe atoms into SnO2, the magnetic behavior of semiconductor changed from diamagnetic to ferromagnetic. It may be due to substitution of Fe atoms in Sn site in SnO2 lattice. The saturation magnetization increased with an increase of Fe doping concentration and became high enough for the sample at a doping concentration of x=0.07. The samples exhibited a magnetization of 95.99 memu/g at Fe doping concentration of x=0.03 and a highest of magnetization 151.41 memu/g at a Fe dopant concentration of x=0.07 was observed. After that, it again decreased even though it had Fe2O3 phases at higher dopant concentrations. When the Fe doping concentration further increased, the saturation magnetic moment decreased. From the X-ray diffraction, it is clear that when Fe doping concentration increased to x=0.15, Fe2O3 along with SnO phase was also observed. But the saturation magnetic moment did not increase even Fe2O3 phase was present in the sample. It reflects that the ferromagnetism is not due to Fe2O3 clusters where as it is an intrinsic property rather than impurities. This confirms that the observed ferromagnetism in Sn1−x Fe x O2 powder samples at x=0.03, 0.05, 0.07 are purely intrinsic in nature. It may be due to ferromagnetic interactions developed in the samples at lower dopant concentrations. Ferromagnetism was also observed in noncrystalline Fe doped SnO2 due to presence of secondary phases [34]. The decrease in magnetic moment at higher Fe doping concentration was observed, which may be due to antiferromagnetic super exchange interaction among the nearest neighbor in Fe atoms. The observed magnetic moments are higher when compared to Fe doped SnO2 at a doping concentration of x=0.10. From Fig. 6(c), it can be seen that the M–H curves are along the origin as the observed magnetizations are very small. The observed magnetizations are equal to that of pure SnO2.

(a) M–H curve of Sn1−x Fe x O2 powder sample at x=0.07. (b) M–H curve Sn1−x Fe x O2 powder sample at x=0.15. (c) M–H curves of Sn1−x Fe x O2 powder samples at x=0.00, 0.03, 0.05, 0.07, and 0.15
4 Conclusions
Iron-doped SnO2 diluted magnetic semiconducting powders (Sn1−x Fe x O2, x=0.00, 0.03, 0.05, 0.07, 0.10, and 0.15) were synthesized by a simple solid state reaction followed by vacuum annealing and studied the effect of Fe doping concentration on structural, optical, and ferromagnetic properties of the synthesized powder samples. The powder samples exhibited tetragonal structure at lower Fe dopant concentrations. At higher dopant concentrations, in addition to tetragonal SnO2, SnO and Fe2O3 phases were also observed. The samples at lower and higher Fe dopant concentrations exhibited ferromagnetism at room temperature and a decrease in magnetic moment was observed at higher Fe doping concentrations.
References
Dietl, T., Ohno, H., Mastukura, F., Cibert, J., Ferrand, D.: Science 287, 1019–1022 (2000)
Ando, K., Saito, H., Zayets, V., Debnath, M.C.: J. Phys. Condens. Matter 16, S5541–S5548 (2004)
Akai, H.: Phys. Rev. Lett. 81, 3002–3005 (1998)
Ohno, H.: J. Magn. Magn. Mater. 200, 110–129 (1999)
Ohno, H.: J. Vac. Sci. Technol., B 18, 2039–2043 (2000)
Wolf, S.A., Awschalom, D.D., Buhrman, R.A., Daughton, J.M., Von Molnar, S., Roukes, M.L., Chtchelkanova, A.Y., Treger, D.M.: Science 294, 1488–1495 (2001)
Chen, Z.W., Li, H.J., Jiao, Z., Wu, M.H., Shek, C.H., Wu, C.M.L., Lai, J.K.L.: Acta Mater. 57, 5078–5082 (2009)
Mizokawa, T., Nambu, A., Fujimori, T., Fukumura, T., Kawasaki, M.: Phys. Rev. B 65, 0852091 (2002)
Matsumoto, Y., Murakami, M., Shono, T., Hasegawa, T., Fukumura, T., Kawasaki, M., Ahmet, P., Chikyow, T., Koshihara, S., Koinuma, H.: Science 291, 854–856 (2001)
Punnoose, A., Seehra, M.S., Park, W.K., Moodera, J.S.: J. Appl. Phys. 93, 7867–7870 (2003)
Coey, J.M.D., Douvalis, A.P., Fitzgerald, C.B., Venkatesan, M.: Appl. Phys. Lett. 84, 1332–1334 (2004)
Fitzgerald, C.B., Venkatesan, M., Dorneles, L.S., Gunning, R., Stamenov, P., Coey, J.M.D., Stampe, P.A., Kennedy, R.J., Moreira, E.C., Sias, U.S.: Phys. Rev. B 74, 115307 (2006)
Jiang, F.X., Xu, X.H., Zhang, J., Fan, X.C., Wu, H.S., Gehring, G.A.: Appl. Phys. Lett. 96, 052503 (2010)
Singhal, R.K., Samariya, A., Kumar, S., Sharma, S.C., Xing, Y.T., Deshpande, U.P., Shripathi, T., Saitovitch, E.: Appl. Surf. Sci. 257, 1053–1057 (2010)
Behan, A.J., Mokhtari, A., Blythe, H.J., Score, D., Xu, X.H., Neal, J.R., Fox, A.M., Gehring, G.A.: Phys. Rev. Lett. 100, 047206 (2008)
Xu, X.H., Blythe, H.J., Ziese, M., Behan, A.J., Neal, J.R., Mokhtari, A., Ibrahim, R.M., Fox, A.M., Gehring, G.A.: New J. Phys. 8, 135–146 (2006)
Qin, H., Zhang, Z., Liu, X., Zhang, Y., Hu, J.: J. Magn. Magn. Mater. 322, 1994–1998 (2010)
Prakash, R., Kumar, S., Ahmed, F., Lee, C.G., Song, J.: Thin Solid Films 519, 8243–8246 (2011)
Hong, N.H.: J. Magn. Magn. Mater. 303, 338–343 (2006)
Fitzgerald, C.B., Venkatesan, M., Douvalis, A.P., Huber, S., Coey, J.M.D.: J. Appl. Phys. 95, 7390–7392 (2004)
Sharma, A., Varshney, M., Kumar, S., Verma, K.D., Kumar, R.: Nanosyst. Nanomater. Nanotechnol. 1, 24–28 (2011)
Sanchez, L.C., Calle, A.M., Arboleda, J.D., Osorio, J., Nomura, K., Barrero, C.A.: Microelectron. J. 39, 1320–1321 (2008)
Torres, C.E.R., Cabrera, A.F., Sanchez, F.H.: Physica B 389, 176–179 (2007)
Cabrera, A.F., Navarro, A.M.M., Torres, C.E.R., Sanchez, F.H.: Physica B 398, 215–218 (2007)
Sambasivam, S., Chun Choi, B., Lin, J.G.: J. Solid State Chem. 184, 199–203 (2011)
Mishra, A.K., Sinha, T.P., Bandyopadhyay, S., Das, D.: Mater. Chem. Phys. 125, 252–256 (2011)
Castro, R.H.R., Pereira, G.J., Gouve, D.: Appl. Surf. Sci. 253, 4581–4585 (2007)
Mathew, X., Enriquez, J.P., Garcia, C.M., Puente, G.C., Jacome, M.A., Antonio, J.A.T., Hays, J., Punnoose, A.: J. Appl. Phys. 100, 073907 (2006)
Cullity, B.D.: Elements of X-ray Diffraction, 2nd edn. p. 102. Addison Wesley, Reading (1978)
Lacombe, S., Cardy, H., Soggiu, N., Blanc, S., Jiawan, J.L.H., Soumillion, J.P.: Microporous Mesoporous Mater. 46, 311–325 (2001)
Tauc, J.: Amorphous and Liquid Semiconductors. Plenum Press, New York (1974)
Abdel Hakeem, A.M.: J. Magn. Magn. Mater. 324, 95–99 (2012)
Hong, N.H., Sakai, J., Huong, N.T., Poirot, N., Ruyter, A.: Phys. Rev. B 72, 045336 (2005)
Sakuma, J., Nomura, K., Barrero, C., Takeda, M.: Thin Solid Films 515, 8653–8655 (2007)
Acknowledgements
Authors are highly thankful to VIT-SIF for providing XRD, DRS facilities to carry out the present work. The authors also thank the Sophisticated Advanced Instruments Facility (SAIF), IIT Madras, Tamilnadu, India, for providing vibrating sample magnetometer facility.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Kuppan, M., Kaleemulla, S., Madhusudhana Rao, N. et al. Physical Properties of Sn1−x Fe x O2 Powders Using Solid State Reaction. J Supercond Nov Magn 27, 1315–1321 (2014). https://doi.org/10.1007/s10948-013-2457-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-013-2457-0