
Abstract
Transforming growth factor-β/bone morphogenetic protein (TGF-β/BMP) signaling plays a fundamental role in embryonic skeletal development and postnatal bone homeostasis. The signaling pivot protein BMP-2 belongs to the TGF-β superfamily and has been implicated in the pathogenesis of osteonecrosis of femoral head (ONFH). The biologically functional BMP-2 is a homodimer that has two tightly packed cores at its dimerization interface; each core is defined by the intermolecular interaction between a helical arm from one monomer and a hydrophobic pocket from another monomer. Inhibition and disruption of BMP-2 dimerization have been recognized as an attractive therapeutic strategy against ONFH. Here, we investigate the self-binding behavior of helical arm-derived peptides to the BMP-2 dimerization interface. The native BMP-2 helical arm and its several grafted versions from BMP-4, BMP-6 and BMP-7 are stripped from the intact dimerization interface to generate a number of isolated helical peptides. Computational simulations demonstrate that the stripping does not substantially influence the direct intermolecular interaction between BMP-2 monomer and these helical peptides or desolvation effect upon the interaction. However, the C-terminus of stripped peptides is found to have an intrinsic disorder and large flexibility in the isolated state, which would impair the rebinding of stripped peptides to BMP-2. Next, we rationally design a hydrocarbon bridge across the C-terminal residues 65 and 69 of helical peptides, which can effectively constrain peptide conformational flexibility in the isolated state, thus considerably promoting the binding potency of stripped helical peptides. Circular dichroism (CD) spectroscopy reveals that the peptide helicity increases from 51.8 to 67.9% upon hydrocarbon stapling. Fluorescence polarization assays substantiate that, as designed, the stapling can convert these helical peptides from weak binders to moderate or good binders of BMP-2 protein; their Kd values are improved by up to ~ fourfold.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Transforming growth factor-β/bone morphogenetic protein (TGF-β/BMP) signaling has widely recognized roles in osteoblast differentiation during mammalian development and exhibits versatile regulatory functions in the bone system [1, 2]. The signaling involves a number of bone morphogenetic proteins (BMPs), which are a group of growth factors that belong to the TGF-β superfamily of cytokines and metabologens. Similar to other TGF-β family proteins, BMPs are highly conserved across animal species [3] and have a central role in the regulation of bone induction, maintenance and repair. They act through an autocrine or paracrine mechanism by binding to cell surface receptors and initiating a cascading of downstream cell signaling events that have multiple effects on the formation of bone and cartilage [4]. In the BMP family, BMP-2 is one of the most documented members because of its functional importance and clinical significance. The protein is a central regulator of bone defects, non-union fractures, spinal fusion, and osteoporosis [5], and has therapeutic benefits for osteonecrosis of femoral head (ONFH) through induction of cartilage and bone cells [6]. Combined therapy of BMP-2 with Ibandronate preserves the shape of the femoral head and stimulates new bone formation in an immature animal model of ONFH [7]. Recombinant human BMP-2 can improve the clinical efficacy and quality of bone repair in debridement and bone graft for the treatment of ONFH [8].
Several forms of BMP-2 exist: a mature active 30 kDa homodimer, an N-terminal propeptide of 40–45 kDa, and a small amount of 60 kDa precursor protein [9]. Proteolytic hydrolysis of the precursor protein produces variable-length propeptides, which can be further cleaved to the mature homodimer. The mature BMP-2 dimer has a large hydrophobic surface exposed to solvent, contributing to its unusually low solubility in aqueous solutions [10]. The BMP-2 monomer can also form a series of functionally active heterodimers with other members of this family, such as BMP-2/6 and -2/7 [11, 12]. Crystallographic analysis revealed that the BMP-2 homodimer possesses two tightly packed cores at its dimerization interface; each core is defined by the intermolecular interaction between a helical arm from one monomer and a hydrophobic pocket from another monomer. The helix is linked to the rest of its host monomer protein through two highly flexible loops (i.e. N-terminal and C-terminal linkers), thus possessing a considerable independence (Fig. 1) [13]. Consequently, the BMP-2 dimerization can be regarded as a peptide-mediated protein–protein interaction (PmPPI) [14, 15].

Crystal structure of BMP-2 homodimer (PDB: 3BMP). Two packed cores can be observed at the dimerization interface. Each core is defined by the intermolecular interaction between a helical arm from one monomer and a hydrophobic pocket from another monomer. The helix is linked to rest of its host monomer protein through two highly flexible loops, i.e. the N-terminal and C-terminal linkers. The monomers 1 and 2 are colored in purple and green, respectively. (Color figure online)
Previously, we have described a protocol to derive self-inhibitory peptides (SIPs) from the crystal complex interface of TGF-β with its cognate receptor TβRI in the ONFH TGF-β/BMP signaling [16]. The protocol integrates computational peptide design [17, 18] and the results from in vitro binding assays to identify, modify, and cyclize the hotspot peptide segments of TβRI that can tightly pack against TGF-β and exert strong interaction potency with the TGF-β. In this study, we performed a systematic investigation of the molecular basis and conformational properties of helical peptide in BMP-2 dimerization. We also attempted to structurally graft the helical peptide counterparts from BMP-X to the BMP-2 to mimic the BMP-2/BMP-X heterodimerization. The native and grafted helical peptides were stripped from their host protein context, stapled by an all-hydrocarbon bridge to stabilize the helical conformation, and tested in vitro to determine their binding potency to BMP-2 monomer. The grafted, stripped and stapled peptides may be exploited as potential biologic agents to regulate the BMP-2 dimerization and would be used as lead molecular entities to develop therapeutic peptide drugs against ONFH and other bone diseases.
2 Materials and Methods
2.1 Computational Modeling
The crystal structure of BMP-2 homodimer was retrieved from the PDB database [19] with entry code 3BMP (Fig. 1). The investigated system was immersed into a TIP3P water box [20] with 10 Å buffer extension. Counterions were added to make the system electroneutral. The studied systems were modeled with AMBER force field [21] implemented in Amber suite of programs [22]. For hydrocarbon-stapled peptides, the stapled alkane bridges were modeled using the XLEAP module and the parameters were built using the ANTECHAMBER module [23].
2.1.1 Dynamics
The system was relaxed by 1000 steps of steepest descent minimization and 5000 steps of conjugate gradient minimization. The energy minimization was followed by heating from 0 to 300 K over 50 ps in the canonical ensemble and by equilibrating to adjust the solvent density under 1 atm pressure over 50 ps in the isothermal isobaric ensemble [24,25,26]. Subsequently, after a 10-ns simulation for relaxing and equilibration, an additional 20-ns production simulation was run, during which conformational snapshots were collected every 10 ps [27]. No extra restraints were set for solute. The time step was 2 fs. All hydrogen-involved chemical bonds were constrained using the SHAKE algorithm [28]. Nonbonded cutoff was set at 10 Å and long-range electrostatic interactions were described with the particle mesh Ewald (PME) method [29]. The structural minimizations and dynamics simulations were carried out using the SANDER module.
2.1.2 Energetics
Molecular mechanics/Poisson–Boltzmann surface area (MM/PBSA) method [30] was carried out over collected snapshots to analyze the binding energetics of BMP-2 dimerization or BMP-2–peptide interaction. The method calculated intermolecular interaction energy ΔGMM between the dimerizing/interacting members using molecular mechanics approach, as well as the desolvation energy ΔGPBSA upon the dimerization/interaction by the finite-difference solution of Poisson–Boltzmann equation and surface area model [31,32,33]. Consequently, the total binding free energy ΔGTTL of dimerization or interaction is expressed as: ΔGTTL = ΔEMM + ΔGPBSA, where ΔEMM is the interaction potential between protein and peptide, and ΔGPBSA is the desolvation energy upon the interaction. The energetics calculation was conducted using the MM/PBSA module. Entropy effect (–TΔS) was not explicitly considered in the calculations.
2.2 Experimental Analysis
2.2.1 Material Preparation
Peptides were synthesized by Fmoc-solid phase chemistry in ChPept. The Grubbs catalyst was used in olefin metathesis to incorporate the all-hydrocarbon staples into peptides, which entailed the integration of two (i,i + 4)-spaced residues followed by ruthenium-mediated olefin metathesis before cleavage from the synthesis resin and deprotection, to yield the stapled peptides [34, 35]. The recombinant protein of human BMP-2 is natively in homodimer form stabilized by a disulfide bond (Cys78-Cys78) across two monomers, which was treated with 100 mM DTT at 37 °C for 30 min to prepare the monomer form of BMP-2 [36]. After then, 25% HSA was added to absorb excessive reducing equivalents [37].
2.2.2 CD Spectroscopy
The circular dichroism (CD) analysis was performed at 25 °C on a Jasco J-810 spectropolarimeter using a path length of 1 mm. All CD signals were recorded at a peptide concentration 50 µM in 10 mM phosphate buffer (pH 7.4). Data acquisition was performed in steps of 0.5 nm at a wavelength range from 180 to 250 nm with a spectral bandwidth of 1.0 nm, and the average blank spectra were subtracted from the average of the sample spectra. The final spectra were expressed as molar ellipticity [θ] (deg cm2 dmol−1) per residue. The α-helical content (%Helicity) was determined from the mean residue ellipticities at 222 nm, as indicated by [38, 39]: %Helicity = ([θ]obs × 100)/{[θ]helix × (1–2.57/l)}, where [θ]obs is the mean residue ellipticity (MRE) observed at 222 nm, [θ]helix is the ellipticity of a peptide of infinite length with a 100% helix, taken as − 39,500 deg cm2 dmol−1, and l is the peptide length or, more precisely, the number of peptide bonds.
2.2.3 Fluorescence Polarization
The binding affinity of peptide ligands to BMP-2 monomer protein was measured by fluorescence polarization (FP) assays modified from our previous work [16]. Saturation binding curves for the BMP-2–peptide interaction were determined by monitoring the FP change of a fixed concentration (100 nM) of labeled peptide with increasing amounts of the protein at room temperature (25 °C) in a buffer 25 mM HEPES, pH 7.4, 0.1% Tween 20, 50 mM NaCl and 10 mM DTT. The dissociation constants (Kd) were determined by fitting titration curves to the equation: FP = {FP0 + FPmax([p]/Kd)}/{1 + ([p]/Kd)}, where [p] is the protein concentration at each measurement point; FP is measured polarization at the given protein concentration; FP0 and FPmax are the polarization in absence of protein and saturated with protein, respectively.
3 Results and Discussion
3.1 Computational Alanine Scanning Analysis of BMP-2 Dimerization
The mature BMP-2 is a homodimer with one disulfide bond across the dimerization interface. However, formation of the disulfide bond is a very slow process [40], before which the two BMP-2 monomers should be fully structured and then properly match to each other at the dimerization interface. Therefore, design of peptide ligands to disrupt the slow process of the folding, binding and matching between two BMP-2 monomers before the disulfide bond formation can potentially be used for therapeutic purpose. The crystal structure of BMP-2 homodimer was subjected to a 20-ns MD simulation for equilibrium and an additional 20-ns simulation for trajectory analysis. In fact, the first 20-ns simulation is sufficient to stabilize the system since the starting crystal structure is already near its energy minimum. Cheminformatics and molecular docking have been widely used to study protein–peptide binding [41, 42]. The total binding energy ΔGTTL of BMP-2 dimerization was calculated as − 18.7 kcal/mol by MM/PBSA analysis of the additional 20-ns dynamics trajectory, indicating that the two monomers can pack against each other to form a stable dimer complex system. Here, computational alanine scanning (CAS) [43] was conducted to determine the residue importance of a monomer in the dimerization. The CAS separately mutated each residue of a monomer in BMP-2 homodimer to neutral alanine and then calculated change in the total dimerization energy ΔΔGTTL upon the mutation, and the obtained ΔΔGTTL value can be used to measure the relative importance of each monomer residue in dimerization; a residue that favorably and unfavorably contributes to the dimerization is labeled with ΔΔGTTL > 0 and ΔΔGTTL < 0, respectively. As shown in Fig. 2, most residues contribute favorably to the dimerization. This is not unexpected if considering that the BMP-2 homodimer is evolutionally polished to obtain a high complementarity at its dimerization interface, so that the CAS mutation would impair the complementarity and then cause unfavorable effects on the formation of the dimer complex (ΔΔGTTL > 0). However, most mutations only influence the dimerization modestly, with ΔΔGTTL < 1 kcal/mol. This is because only a few interfacial residues can directly participate in the dimerization interaction, while a majority of residues are non-interfacial and can only exert indirect effects on the interaction [44, 45]. Visual examination of the dimer complex structure revealed that a number of important residues are clustered into a hotspot region at the dimerization interface, which roughly covers an α-helical arm of BMP-2 monomer and just corresponds to one of the two packed cores in the interface, where the helix is linked to the rest of the monomer protein through two highly flexible loops (Fig. 2). The helical arm spans the residues 58–71 of BMP-2 protein sequence and defines a functional peptide 58TNHAIVQTLVNSVN71. According to energetics analysis the single peptide segment can contribute 35.8% binding energy to BMP-2 dimerization, suggesting that the BMP-2 dimerization is a typical peptide-mediated protein–protein interaction (PmPPI) and the helical peptide is the central mediator of the dimerization interaction.

Computational alanine scanning determination of the residue importance profile (ΔΔGTTL) of a monomer in BMP-2 dimerization. The monomer consists of 106 residues (residues 9–114) in investigated crystal structure (PDB: 3BMP)
3.2 Grafting Helical Arms from BMP-4/6/7 to BMP-2 Dimer
Although BMP-2 commonly exists in homodimer form, in some cases the protein can also form biologically functional heterodimers with BMP-4 [46], BMP-6 [11] and BMP-7 [12]. These heterodimers have been found as strong inductors of osteoblastic differentiation and bone regeneration in human embryonic stem cells and rat spinal fusion (Table 1). Considering that the BMP family proteins are highly conserved and share a high sequence homology and structural similarity, the packed cores at heterodimerization interface should also be highly consistent with that in BMP-2 homodimer, where the helical arm of one monomer tightly packs against a hydrophobic pocket of another monomer. In this respect, we separately grafted the helical arms of BMP-4, BMP-6 and BMP-7 to BMP-2 homodimer; it is supposed that the grafting would not influence the conformation and affinity of dimerization substantially.
First, the primary sequence of BMP-2, BMP-4, BMP-6 and BMP-7 were retrieved from the UniProt database [47] and summarized in Table 1, which were then compared with each other using ESPript multiple sequence alignment (MSA) [48]. In addition, the helical arm is also highly consistent across the four BMP proteins, in which only three variable residues are observed (Fig. 3). In fact, the helical arm sequences of BMP-2 and BMP-4 are identical. Second, the BMP-6 and BMP-7 helical arms were grafted to the packed cores of BMP-2 homodimer using a virtual mutagenesis strategy. In the procedure, the side chains of different residues in the two helical arms of BMP-2 homodimer crystal structure were manually removed and new side chains were then added to these residues using the rotamer-based SCWRL4 program [49], consequently resulting in the BMP-6 and BMP-7 helical arm-grafted versions of BMP-2 dimer structure. Subsequently, the two grafted structures were separately subjected to 40-ns MD simulations (first 20-ns for equilibrium and last 20-ns for trajectory analysis). As might expected, the dimerization potency has not been influenced considerably upon the grafting, with total binding energy ΔGTTL change from − 18.7 kcal/mol (native) to − 16.8 (BMP-6) and − 17.3 (BMP-7) kcal/mol, respectively (Table 1). In fact, both the native BMP-2 and grafted BMP-6/BMP-7 helical arms are very similar, and no obvious change in their conformation and packing mode can be observed in the MD-equilibrated dimer structures.

Multiple sequence alignment of BMP-2, BMP-4, BMP-6 and BMP-7. The helical arm region is highlighted in a box, where three variable residues are identified across the four proteins. The alignment was carried out using ESPript server [48]. The conserved and identical residues are highlighted by yellow and red, respectively. (Color figure online)
3.3 Stripping and Stapling of BMP-2-Derived Helical Peptides
The helical arm was stripped from the crystal structure of BMP-2 homodimer to derive the isolated B2 peptide (Fig. 4a), which was then subjected to 50-ns MD simulations to reach equilibrium (Fig. 4b). As can be seen, the equilibrated conformation of B2 peptide has a well structured N-terminus where the peptide is folded into helical conformation, whereas the peptide’s C-terminus exhibits an intrinsic disorder and large flexibility. We also performed MD simulations to equilibrate B6 and B7 peptides and a similar conformational dynamics behavior was observed, indicating that these stripped peptides are partially unfolded in the isolated state. Previous study found that flexible peptide ligands would incur an entropy penalty when binding to their protein receptors, which considerably impairs the apparent binding capability of protein–peptide interactions [50].

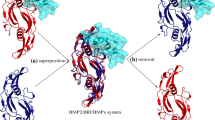
Schematic representation of peptide flexibility reduction upon hydrocarbon stapling. a The helical arm is stripped from BMP-2 homodimer interface to derive an isolated B2 peptide. b The native helical conformation of B2 peptide is subjected to 50-ns MD simulations. The equilibrated peptide possesses a structured N-terminus, but exhibits a large intrinsic disorder in its C-terminus. c The B2 peptide is stapled by a hydrocarbon bridge across residues 65 (i) and 69 (i + 4), and then subjected to 50-ns MD simulations. The equilibrated conformation of stapled peptide is highly structured, fully holding a helical conformation. d CD spectra of B2 and sB2 peptides with their respective secondary structures
To reduce the conformational flexibility of stripped B2, B6 and B7 peptides, hydrocarbon stapling was used to constrain these peptides in a helical conformation [51]. In order to avoid disruption of the native interaction of BMP-2 protein with these helical peptides, the hydrocarbon bridge should be placed on the side of the helix facing away from the binding pocket [35]. In addition, considering that the C-terminus of the helix is disordered in the isolated state, we stapled a hydrocarbon bridge in the flexible terminus. By visual examination of BMP-2-helical arm binding mode we selected peptide residues 65 (i) and 69 (i + 4) as the anchor sites of hydrocarbon bridge. This is because the two residues are located near the peptide’s C-terminus, do not directly contact and interact with BMP-2 protein, and are at the (i, i + 4) positions in the peptide sequence. The stapled B2 peptide (sB2 peptide) was re-subjected to 50-ns MD simulations and, as might be expected, the peptide is highly structured and can be maintained in helical conformation during the whole simulation course (Fig. 4c). Circular dichroism (CD) spectroscopy was also used to characterize the secondary structure of native B2 peptide and stapled sB2 peptide [52]. The two peptides are very similar in sequence, size, mass and amino acid composition, and thus the CD can readily reflect the structural difference between them. As shown in Fig. 4d, both the two peptides exhibit helical propensity, with a positive band at ~ 193 nm and negative bands between 205 and 225 nm—this represents a typical α-helical feature [53]. By comparing the spectral profiles it is revealed that the sB2 peptide has a higher helical content than B2 peptide. In addition, no concentration effects were observed on the shape and intensity of CD spectra in the range (10−4 to 10−5 M) taken into consideration, suggesting that the peptides in tested concentration (50 µM) are not self-associated. Here, the peptide helicity was derived from the CD spectra using a method described previously [38, 39]. In addition, the average helical propensity (AHP) of the two peptides during the equilibrium phase of MD simulations was also calculated, where the peptide secondary structure definition was assigned with the DSSP algorithm [54]. As can be seen in Table 2, the computational simulations suggested that the stapling can increase AHP from 7.8/14 (55.7%) to 12.3/14 (87.9%), which is basically in line with the change in peptide helicity upon the stapling (increase from 51.8 to 67.9%) derived from the experimental CD spectra, although the experimental helicity is generally lower than computational AHP for the two peptides.
For comparison purposes, both the native B2/B6/B7 peptides and their stapled counterparts sB2/sB6/sB7 are listed in Table 3. As might be expected, hydrocarbon stapling does not influence the direct interaction (ΔGMM) and desolvation effect (ΔGPBSA) of these helical peptides binding to BMP-2 protein, with a small variation in total binding energy (ΔGTTL ranges between − 5.8 and − 7.3 kcal/mol). Instead, the stapling can effectively stabilize the helical peptides in the isolated state, thus largely promoting native peptide interaction with BMP-2. Consequently, the apparent binding capability of these peptides is expected to be improved considerably due to the stapling-mediated conformational constraint. Although the binding energy of designed peptides to BMP-2 monomer (− 7 kcal/mol) is about 1/2 or 1/3 of the BMP-2 dimerization energy (− 18 kcal/mol), this is expected as the dimerization interface contains two peptide binding sites (each comes from one monomer and binds to another monomer, see Fig. 1) and thus the total dimerization energy should be regarded as double the binding energy of one single peptide. In addition, since some additional factors such as entropy effects were not involved in the energetic calculations, the binding energy may not be directly comparable between the small peptide and big BMP-2 protein. Here, considering that these investigated peptides have the same length (14-mer) and share a similar sequence (only three residues difference), the entropy penalty (− TΔS) upon binding to their common protein receptor BMP-2 can be regarded as a constant over three unstapled peptides (B2, B6 and B7) or over three unstapled peptides (sB2, sB6 and sB7), and thus the relative values of their calculated binding energies (ΔGTTL) are comparable with each other, albeit the absolute ΔGTTL values may not be reliable due to the lack of − TΔS. Moreover, he mature BMP-2 is a homodimer with one disulfide bond across the dimerization interface. However, formation of the disulfide bond is a very slow process [40], before which the two BMP-2 monomers should be well structured and then properly match to each other at the dimerization interface. Therefore, the stapled peptides are designed to disrupt the slow process of the folding, binding and matching between two BMP-2 monomers before the disulfide bond formed.
Subsequently, the B2, B6 and B7 peptides as well as their conformationally constrained counterparts sB2, sB6 and sB7 were chemically synthesized and their binding affinity towards BMP-2 monomer was tested using FP assays. As expected, the three native peptides were determined to have only a moderate potency for BMP-2, with Kd values of 78, 156 and 108 µM, respectively. In contrast, hydrocarbon stapling confers a higher affinity for the three peptides, with Kd improved to 21, 62 and 43 µM, respectively (Table 3). For example, the binding curves of native B2 peptide and its stapled counterpart are plotted in Fig. 5. As can be seen, the stapling-mediated conformational constraint of B2 peptide can effectively shift fluorescence polarization associated with the peptide binding to BMP-2, with affinity increase by ~ fourfold (Kd value decreases from 78 to 21 µM). The difference in Kd values obtained for the conformationally constrained (stapled) peptide and the more flexible (unstapled) peptide can be used to make a rough estimate of entropy contribution using the Gibbs relationship: − TΔΔS ≈ ΔΔG = RTln[Kd(stapled)/Kd(unstapled)], where the R is a constant 1.986 × 10−3 kcal K−1 mol−1, and T is the absolute temperature 298.15 K at experimental condition (25 °C). The estimated –TΔΔS values for stapling B2, B6 and B7 peptides (to sB2, sB6 and sB7 peptides, respectively) are − 0.78, − 0.56 and − 0.54 kcal/mol, which cause the peptide affinity improvement by 3.7-fold, 2.6-fold and 2.5-fold, respectively.

Fluorescence polarization binding curves upon titration of BMP-2 protein to labeled peptides B2 and sB2. The titration was determined by monitoring the FP change of a fixed concentration (100 nM) of peptide with increasing amounts of the protein at room temperature (25 °C). The dissociation constants (Kd) were determined by fitting titration curves to the equation: FP = {FP0 + FPmax([p]/Kd)}/{1 + ([p]/Kd)}, where [p] is the protein concentration at each measurement point; FP is measured polarization at the given protein concentration; FP0 (fixed to 80.5 and 81.4 mP for native and stapled peptides, respectively) and FPmax (fixed to 129.2 and 153.6 mP for native and stapled peptides, respectively) are the polarization in absence of protein and saturated with protein, respectively
The modeled structure of hydrocarbon-stapled sB2 peptide in complex with BMP-2 is shown in Fig. 6a. As designed, the hydrocarbon bridge is not at the complex interface and thus should not disrupt the complex interaction. Superposition between the equilibrated structures of native B2 peptide and stapled sB2 peptide in the binding pocket of BMP-2 is shown in Fig. 6b. As can be seen, the binding modes of B2 and sB2 peptides are similar with root-mean-squares deviation (RMSD) of only 0.31 Å; both of them lay across the hydrophobic pocket of BMP-2 and tightly pack against the protein receptor, suggesting that the stapled peptide can bind in a very similar manner as its native counterpart, the helical arm in BMP-2 homodimer. The slight difference between the two binding modes could be (partially) attributed to the bias introduced by computational modeling and MD simulations. In fact, the B2 and sB2 peptides were calculated to have a similar binding energy (ΔGTTL = − 7.3 and − 7.2 kcal/mol, respectively). By visually examining the equilibrated complex structure using the 2D-GraLab program [55], it is revealed that the stapled sB2 peptide can form a variety of noncovalent interactions with BMP-2, including three hydrogen bonds and a number of hydrophobic interactions and van der Waals contacts (Fig. 6c). These noncovalent interactions are mostly distributed at the peptide residues that face and directly contact the protein, thus displaying an alternate pattern along the primary sequence of the helical peptide and conferring high stability and moderate specificity to the protein–peptide binding.

a The computationally modeled, dynamics-equilibrated structure of BMP-2 monomer in complex with hydrocarbon-stapled sB2 peptide. As designed, the hydrocarbon bridge is not presented at the complex interface and thus should not disrupt the complex interaction. b Superposition between the equilibrated structures of native B2 peptide (in intact interface context) and hydrocarbon-stapled sB2 peptide in the binding pocket of BMP-2. The binding modes of B2 and sB2 peptides are similar with RMSD = 0.31 Å; both of them lay across the hydrophobic pocket of BMP-2 and tightly pack against the protein receptor, suggesting that the stapled peptide can bind in a very similar manner as its native counterpart of helical arm in the BMP-2 homodimer. c Schematic representation of noncovalent interactions across the BMP-2–sB2 peptide complex interface. The stapled sB2 peptide forms a variety of noncovalent interactions with BMP-2, including three hydrogen bonds and a number of hydrophobic forces and van der Waals contacts. These chemical forces are mostly distributed at the peptide residues that face and directly contact the protein, thus displaying an alternate pattern along the primary sequence of the helical peptide and conferring high stability and moderate specificity to the protein–peptide binding. The plot was generated using 2D-GraLab program [55]
4 Conclusions
Helical peptide segments were stripped from the homo- and hetero-dimerization interface of BMP-2 with BMP-2, BMP-6 and BMP-7. These isolated peptides cannot be fully maintained in native helical conformation; they have an increased flexibility and intrinsic disorder without the support of the interface context, thus impairing their rebinding capability for targeting the dimerization. Stapling strategy was employed to constrain the helical conformation of peptides by chemically adding a hydrocarbon bridge across the peptide C-terminal (i, i + 4) residues, which considerably reduces the peptide flexibility and helps them to interact with BMP-2, with affinity increase by 2–4-fold. The hydrocarbon bridge is placed on the side of the helix facing away from the peptide-binding pocket and hence would not disrupt the interaction. The native and stapled peptides can bind to BMP-2 in a similar manner; both of them lay across the hydrophobic pocket of BMP-2 and pack tightly against the protein.
References
Chen G, Deng C, Li YP (2012) TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int J Biol Sci 8:272–288
Wu M, Chen G, Li YP (2016) TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res 4:16009
Hogan BL (1996) Bone morphogenetic proteins: multifunctional regulators of vertebrate development. Genes Dev 10:1580–1594
Sykaras N, Opperman LA (2003) Bone morphogenetic proteins (BMPs): how do they function and what can they offer the clinician? J Oral Sci 45:57–73
Chen D, Zhao M, Mundy GR (2004) Bone morphogenetic proteins. Growth Factors 22:233–241
Wang C, Zang H, Zhou D (2018) Bone morphogenetic protein-2 exhibits therapeutic benefits for osteonecrosis of the femoral head through induction of cartilage and bone cells. Exp Ther Med 15:4298–4308
Vandermeer JS, Kamiya N, Aya-ay J, Garces A, Browne R, Kim HK (2011) Local administration of ibandronate and bone morphogenetic protein-2 after ischemic osteonecrosis of the immature femoral head: a combined therapy that stimulates bone formation and decreases femoral head deformity. J Bone Joint Surg Am 93:905–913
Sun W, Li Z, Gao F, Shi Z, Zhang Q, Guo W (2004) Recombinant human bone morphogenetic protein-2 in debridement and impacted bone graft for the treatment of femoral head osteonecrosis. PLoS ONE 9:e100424
Israel DI, Nove J, Kerns KM, Moutsatsos IK, Kaufman RJ (1992) Expression and characterization of bone morphogenetic protein-2 in Chinese hamster ovary cells. Growth Factors 7:139–150
Ruppert R, Hoffmann E, Sebald W (1996) Human bone morphogenetic protein 2 contains a heparin-binding site which modifies its biological activity. Eur J Biochem 237:295–302
Valera E, Isaacs MJ, Kawakami Y, Izpisúa Belmonte JC, Choe S (2010) BMP-2/6 heterodimer is more effective than BMP-2 or BMP-6 homodimers as inductor of differentiation of human embryonic stem cells. PLoS ONE 5:e11167
Morimoto T, Kaito T, Matsuo Y, Sugiura T, Kashii M, Makino T, Iwasaki M, Yoshikawa H (2015) The bone morphogenetic protein-2/7 heterodimer is a stronger inducer of bone regeneration than the individual homodimers in a rat spinal fusion model. Spine J 15:1379–1390
Scheufler C, Sebald W, Hülsmeyer M (1999) Crystal structure of human bone morphogenetic protein-2 at 2.7 Å resolution. J Mol Biol 287:103–115
Petsalaki E, Russell RB (2008) Peptide-mediated interactions in biological systems: new discoveries and applications. Curr Opin Biotechnol 19:344–350
Zhou P, Hou S, Bai Z, Li Z, Wang H, Chen Z, Meng Y (2018) Disrupting the intramolecular interaction between proto-oncogene c-Src SH3 domain and its self-binding peptide PPII with rationally designed peptide ligands. Artif Cells Nanomed Biotechnol 46:1122–1131
Zhu Z, Zhang C, Song W (2017) Rational derivation, extension, and cyclization of self-inhibitory peptides to target TGF-β/BMP signaling in ONFH. Amino Acids 49:283–290
Zhou P, Wang C, Ren Y, Yang C, Tian F (2013) Computational peptidology: a new and promising approach to therapeutic peptide design. Curr Med Chem 20:1985–1996
Zhou P, Yang C, Ren Y, Wang C, Tian F (2013) What are the ideal properties for functional food peptides with antihypertensive effect? A computational peptidology approach. Food Chem 141:2967–2973
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW (1983) Comparison of simple potential functions for simulating liquid water. J Chem Phys 79:926–935
Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T, Caldwell J, Wang J, Kollman P (2003) A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 24:1999–2012
Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ (2005) The Amber biomolecular simulation programs. J Comput Chem 26:1668–1688
Joseph TL, Lane DP, Verma CS (2012) Stapled BH3 peptides against MCL-1: mechanism and design using atomistic simulations. PLoS ONE 7:e43985
Yang C, Wang C, Zhang S, Huang J, Zhou P (2015) Structural and energetic insights into the intermolecular interaction among human leukocyte antigens, clinical hypersensitive drugs and antigenic peptides. Mol Simul 41:741–751
Yang C, Zhang S, He P, Wang C, Huang J, Zhou P (2015) Self-binding peptides: folding or binding. J Chem Inf Model 55:329–342
Yang C, Zhang S, Bai Z, Hou S, Wu D, Huang J, Zhou P (2016) A two-step binding mechanism for the self-binding peptide recognition of target domains. Mol Biosyst 12:1201–1213
Bai Z, Hou S, Zhang S, Li Z, Zhou P (2017) Targeting self-binding peptides as a novel strategy to regulate protein activity and function: a case study on the proto-oncogene tyrosine protein kinase c-Src. J Chem Inf Model 57:835–845
Ryckaert JP, Ciccotti G, Berendsen HJC (1997) Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 23:327–341
Darden T, York D, Pedersen L (1993) Particale mesh Ewald and N·log(N) method for Ewald sums in large systems. J Chem Phys 98:10089–10092
Homeyer N, Gohlke H (2012) Free energy calculations by the molecular mechanics Poisson-Boltzmann surface area method. Mol Inf 31:114–122
Tian F, Lv Y, Zhou P, Yang L (2011) Characterization of PDZ domain-peptide interactions using an integrated protocol of QM/MM, PB/SA, and CFEA analyses. J Comput Aided Mol Des 25:947–958
Tian F, Tan R, Guo T, Zhou P, Yang L (2013) Fast and reliable prediction of domain-peptide binding affinity using coarse-grained structure models. Biosystems 113:40–49
Tian F, Yang C, Wang C, Guo T, Zhou P (2014) Mutatomics analysis of the systematic thermostability profile of Bacillus subtilis lipase A. J Mol Model 20:2257
Phillips C, Roberts LR, Schade M, Bazin R, Bent A, Davies NL, Moore R, Pannifer AD, Pickford AR, Prior SH, Read CM, Scott A, Brown DG, Xu B, Irving SL (2011) Design and structure of stapled peptides binding to estrogen receptors. J Am Chem Soc 133:9696–9699
Wu T, He P, Wu W, Chen Y, Lv F (2018) Targeting oncogenic transcriptional corepressor Nac1 POZ domain with conformationally constrained peptides by cyclization and stapling. Bioorg Chem 80:1–10
Yano K, Hoshino M, Ohta Y, Manaka T, Naka Y, Imai Y, Sebald W, Takaoka K (2009) Osteoinductive capacity and heat stability of recombinant human bone morphogenetic protein-2 produced by Escherichia coli and dimerized by biochemical processing. J Bone Miner Metab 27:355–363
Lichtenberger FJ, Montague C, Hunter M, Frambach G, Marsh CB (2006) NAC and DTT promote TGF-β1 monomer formation: demonstration of competitive binding. J Inflamm 3:7
Chen YH, Yang JT, Chau KH (1974) Determination of the helix and beta form of proteins in aqueous solution by circular dichroism. Biochemistry 13:3350–3359
Gopal R, Park JS, Seo CH, Park Y (2012) Applications of circular dichroism for structural analysis of gelatin and antimicrobial peptides. Int J Mol Sci 13:3229–3244
Vallejo LF, Rinas U (2013) Folding and dimerization kinetics of bone morphogenetic protein-2, a member of the transforming growth factor-β family. FEBS J 280:83–92
Luo H, Du T, Zhou P, Yang L, Mei H, Ng H, Zhang W, Shu M, Tong W, Shi L, Mendrick DL, Hong H (2015) Molecular docking to identify associations between drugs and class I human leukocyte antigens for predicting idiosyncratic drug reactions. Comb Chem High Throughput Screen 18:296–304
Ren Y, Chen X, Feng M, Wang Q, Zhou P (2011) Gaussian process: a promising approach for the modeling and prediction of Peptide binding affinity to MHC proteins. Protein Pept Lett 18:670–678
Kortemme T, Kim DE, Baker D (2004) Computational alanine scanning of protein–protein interfaces. Sci STKE 2004:pl2
Zhou P, Wang C, Tian F, Ren Y, Yang C, Huang J (2013) Biomacromolecular quantitative structure-activity relationship (BioQSAR): a proof-of-concept study on the modeling, prediction and interpretation of protein–protein binding affinity. J Comput Aided Mol Des 27:67–78
Zhou P, Zhang S, Wang Y, Yang C, Huang J (2016) Structural modeling of HLA-B:1502 peptide carbamazepine T-cell receptor complex architecture: implication for the molecular mechanism of carbamazepine-induced Stevens–Johnson syndrome toxic epidermal necrolysis. J Biomol Struct Dyn 34:1806–1817
Lavery K, Swain P, Falb D, Alaoui-Ismaili MH (2008) BMP-2/4 and BMP-6/7 differentially utilize cell surface receptors to induce osteoblastic differentiation of human bone marrow-derived mesenchymal stem cells. J Biol Chem 283:20948–20958
UniProt C (2015) UniProt: a hub for protein information. Nucleic Acids Res 43:D204–D212
Gouet P, Courcelle E, Stuart DI, Métoz F (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15:305–308
Krivov GG, Shapovalov MV, Dunbrack RL (2009) Improved prediction of protein side-chain conformations with SCWRL4. Proteins 77:778–795
Yu H, Zhou P, Deng M, Shang Z (2014) Indirect readout in protein–peptide recognition: a different story from classical biomolecular recognition. J Chem Inf Model 54:2022–2032
Walensky LD, Bird GH (2014) Hydrocarbon-stapled peptides: principles, practice, and progress. J Med Chem 57:6275–6288
Greenfield NJ (2006) Using circular dichroism spectra to estimate protein secondary structure. Nat Protoc 1:2876–2890
Dotyp H (1965) The ultraviolet circular dichroism of polypeptides. J Am Chem Soc 87:218–228
Kabsch W, Sander C (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22:2577–2637
Zhou P, Tian F, Shang Z (2009) 2D depiction of nonbonding interactions for protein complexes. J Comput Chem 30:940–951
Funding
This work was supported by the National Natural Science Foundation of China (No. 81371962).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Song, W., Wang, K., Wang, W. et al. Grafting, Stripping and Stapling of Helical Peptides from the Dimerization Interface of ONFH-Related Bone Morphogenetic Protein-2. Protein J 38, 12–22 (2019). https://doi.org/10.1007/s10930-018-9807-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10930-018-9807-5