
Abstract
Concerns over gestational effects on the disposition of drugs has highlighted the need for a better understanding of drug distribution and elimination during pregnancy. This study aimed at predicting maternal drug kinetics using a physiologically based pharmacokinetic (PBPK) modelling approach focusing on the observed gestational changes in three important Cytochrome P450 metabolizing enzymes, namely, CYP1A2, CYP2D6 and CYP3A4 at different gestational weeks (GWs). The Pregnancy PBPK model within the Simcyp Simulator V19 was used to predict the pharmacokinetics of sensitive probes to these enzymes; namely caffeine, theophylline, metoprolol, propranolol, paroxetine, midazolam, nifedipine and rilpivirine. PBPK model predictions were compared against clinical data collated from multiple studies for each compound to cover a wide spectrum of gestational ages. Pregnancy PBPK model predictions were within 2-fold error and indicated that CYP1A2 activity is approximately 0.70, 0.44 and 0.30 fold of the non-pregnant level at the end of the first, second and third trimesters, respectively. On the other hand, CYP2D6 activity increases by 1.36, 2.16 and 3.10 fold of the non-pregnant level at the end of the first, second and third trimesters, respectively. Likewise, CYP3A4 activity increases by 1.25, 1.75 and 2.32 fold of the non-pregnant level at the end of the first, second and third trimesters, respectively. The enzymes activity have been qualified throughout pregnancy. Quantified changes in drug dosing are most relevant during the third trimester, especially for drugs that are mainly eliminated by CYP1A2, CYP2D6 and CYP3A4 enzymes. The provided functions describing the continuous changes to the activity of these enzymes during pregnancy are important when modelling long term pharmacokinetic studies where longitudinal modelling or time-varying covariates are used.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pharmacokinetics during pregnancy has gained considerable interest over the last few years. Multifactorial changes develop “physiologically” with pregnancy progression [1] and can alter drugs kinetics [2]. Drug-metabolizing enzyme activity is one of many factors affecting patient exposure and hence response to medications. Among these enzymes, the cytochrome P450 (CYP) enzymes are involved in the metabolism of more than 70% of drugs [3], with CYP3A4 being the most involved enzyme. Other significant enzymes of the CYPs family include CYP2D6, CYP2C9, CYP2C19, CYP1A2, CYP2B6 and CYP2E1 [4]. Genetic variability (polymorphism) in these enzymes are linked to inter-individual differences in drug exposure and response [5]. The CYP2D6 enzyme is highly polymorphic and is involved in the metabolism of a wide range of commonly prescribed drug classes, including antihypertensive agents and antidepressants. Gender-related differences in PK can lead to a different pattern of treatment response in women compared to the typical man due to differences in the activity of major metabolic enzymes, e.g. CYP3A4 and CYP1A2 [6,7,8]. Among these enzymes, the changes to the activity of CYP1A2, CYP2D6 and CYP3A4 during pregnancy have been evaluated in many clinical studies at different gestational weeks (GWs). For example, caffeine clearance in 25 pregnant women was compared with caffeine clearance in the same subjects at 6 to 8 weeks after the delivery, the CYP1A2 activity during pregnancy was reduced from the postpartum level by 33 ± 23%, 48 ± 27%, and 65 ± 15% at 14 to18 GWs, 24 to 28 GWs, and 36 to 40 GWs, respectively [9]. These data are in agreement with another study (n = 12 women) which reported around 35%, 50%, and 52% reduction in CYP1A2 activity at 8–16 GWs, 20–28 GWs and 32–39 GWs, respectively [10].
The activity of CYP2D6 during pregnancy has been reported using either the urinary dextromethorphan/dextrorphan (DEX/DOR) metabolic ratio [9] or measuring the clearance of metoprolol, a drug that is sensitive to CYP2D6 activity [11]. Results from the urinary DEX/DOR metabolic ratio showed that the activity of CYP2D6 increased significantly throughout the pregnancy (26 ± 58% at 14–18 GWs, 35 ± 41% at 24–28 GWs, and 48 ± 25% at 36–40 GWs) as compared with the activity in the same subjects at 6 to 8 weeks postpartum (n = 25) [9]. It is well-known that the urinary DEX/DOR ratio is not a pure marker of CYP2D6 activity as it is affected by the renal function and plasma protein binding [12, 13], which are affected by the gestational age [1]. In another study, using metoprolol as a probe substrate, the clearance in women who were extensive metabolizers to CYP2D6 (EM) increased from 209 ± 92 L/h in non-pregnant women to 432 ± 181 L/h at 22–26 GWs reaching 629 ± 228 L/h at 34–38 GWs [11] suggesting a 2- and 3-fold increase in the activity of CYP2D6 during the second and third trimesters.
Longitudinal changes in CYP3A4 activity during pregnancy have also been reported using the urinary dextromethorphan/3-hydroxymorphinan (DEX/3HM) metabolic ratio [9] from 25 women, measured at 24–28 GWs, and at 36–40 GWs and at 6 to 8 weeks after the delivery. The results showed that CYP3A4 activity was around 35%-38% on both occasions during pregnancy [9]. Midazolam is a sensitive probe of CYP3A4 and its clearance changes during pregnancy have been reported at 28–32 GWs and also at term [14, 15] suggesting an approximately 2-fold higher activity compared to the non-pregnant level, which is higher than those reported using the DEX/3HM metabolic ratio.
The aim of this study was to assess the PBPK prediction at different time points during gestation focusing on the activity changes of CYP1A2, CYP2D6 and CYP3A4 using drugs that are sensitive to these enzymes, namely, caffeine, theophylline, metoprolol, propranolol, paroxetine, midazolam, nifedipine and rilpivirine. These enzymes and compounds were chosen due to data availability. The stochastic PBPK modelling approach that includes gestational-dependent physiology was used to account for inter-individual variability.
Methods
General settings
The pregnancy model within the Simcyp Simulator V19 was used for all predictions in the current study. The model accounts for the known physiological changes that occur during pregnancy, using the Caucasian population data, and allows for the generation of virtual pregnant populations between 0 and 40 GWs. It also accounts for inter-individual variability in the physiological parameters at a specific gestational week [1]. Growth or decline of the physiological parameters during pregnancy are incorporated in the model as continuous functions [16]. The model takes into account the continuous change of all physiological and biological parameters simultaneously over time and within each subject to account for any time-varying covariates. This feature is essential for long term simulations. Previously, a similar approach was introduced and applied in neonate PBPK models [17, 18].
Gestational changes in metabolizing enzymes
CYP1A2 activity
The default equation that describes the activity of CYP1A2 during pregnancy within the Simcyp Simulator was used in this analysis. It describes the continuous fold reduction in the enzyme activity, versus the non-pregnant base line during the whole gestational period (Eq. 1).
where GW is the gestational week. This relationship is developed based on the unbound caffeine clearance using the observed reduction in the total caffeine clearance during pregnancy [9] and correcting the caffeine clearance for the changes in plasma protein binding during pregnancy. Caffeine plasma free fraction (fu) in non-pregnant subjects is around 0.68 and it increases slightly during pregnancy to 0.74 at term due to the fact that the concentration of plasma protein binding is reduced during pregnancy [1]. Equation 1 indicates that the activity of CYP1A2 is about 0.70, 0.44 and 0.30 fold of the non-pregnant level at the end of the first, second and third trimesters, respectively.
CYP2D6 activity
The default equation within the Simcyp Simulator V19 that describes the longitudinal changes in CYP2D6 activity during pregnancy (Eq. 2) was used in this analysis.
The equation is based on the observed changes in the metoprolol clearance in EMs pregnant women [11] and taking into account the change in the binding protein (albumin) during pregnancy. Metoprolol is not highly bound to plasma albumin with an average fu of 0.89 in non-pregnant women increasing slightly during pregnancy to 0.91 towards term [19]. Equation 2 indicates that the activity of CYP2D6 is approximately 1.36, 2.16 and 3.10 fold of the non-pregnant level at the end of the first, second, and third trimesters, respectively.
CYP3A4 activity
The default equation within the Simcyp Simulator V19 that describes the longitudinal changes in CYP3A4 activity during pregnancy (Eq. 3) was used in this analysis as follows:
This equation is based on both the DEX/3HM metabolic ratio [9] and midazolam clearance [14, 15] after correcting observed data for the binding protein. These two metric systems can be integrated due to the fact that midazolam is mainly eliminated via metabolism with almost no renal clearance. In normal healthy subjects the average fu is about 0.035 [20] and increases during pregnancy, due to the reduction in the plasma albumin level [1], reaching to about 0.044 at term. Equation 3 indicates that the CYP3A4 activity is approximately 1.25, 1.75 and 2.32 fold of the non-pregnant level, at the end of the first, second, and third trimesters, respectively.
Model building
For all evaluated compounds, the distribution was defined using a full PBPK distribution model that accounts for different tissue volumes and flow rates. Drugs tissues to plasma partition ratios (Kps) were predicted within the Simulator (see below for more detail). The enzyme kinetics option was used for all drugs in these analyses to describe drug elimination from the body and to allow the scaling of enzyme kinetics of the evolved enzymes from non-pregnant to pregnant women. Where no information was provided on the subject’s phenotypes or genotypes in the original study, the default phenotypes within the Simulator for the general Caucasian population were used.
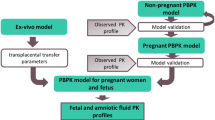
All compound files were first built and verified for their performances in non-pregnant women, and updated if required using non-pregnant data, before they were used to predict the drug kinetics in pregnant women according to the workflow given in Fig. 1. Once the PBPK model predicted the kinetics in non-pregnant women, these settings were retained to predict the drug kinetics in pregnant women at different gestational weeks.

A general workflow for pregnancy PBPK model development
Predictions in non-pregnant and pregnant women were performed via matched clinical settings/designs to the original studies. In all simulations, the number of subjects in the corresponding clinical study were used within each trial. For a better description of the variability, twenty trials were used to simulate virtual subjects. Predicted PK profiles and PK parameters were compared with different sets of clinical observations available in the literature.
Caffeine
Model building
The caffeine compound model within the Simcyp Simulator was used. The default caffeine elimination kinetics, mainly via hepatic CYP1A2, were kept. Caffeine is absorbed quickly and completely from the gut without any gut metabolism. This process was described using the first order absorption model option and was updated to describe plasma concentration in 12 non-pregnant women after administration of 100 mg caffeine three times daily [21]. Caffeine Kps were predicted within the Simulator according to the Rodgers and Rowland method [22] without any modifications.
Caffeine PK during pregnancy
Caffeine pharmacokinetics were studied during the first, second and third trimesters, and concentration profiles being reported in 8 pregnant women at 30–40 GWs and in 4 women on the 4th day after delivery. This study showed an approximate 3-fold increase in the half-life and a 3-fold decrease in the total clearance of caffeine in pregnant women compared with the postpartum results [23]. Other studies also showed an increasing half-life of caffeine with progression of pregnancy between 4 GWs and term [24,25,26,27].
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after the caffeine administration:
Trial design A: Single oral dose of 100 mg caffeine [21]; 20 trials with 12 non-pregnant women in each trial.
Trial design B: Single oral dose of 150 mg caffeine [23]; 20 trials with 4 non-pregnant women in each trial.
Trial design C: Single oral dose of 150 mg caffeine [28]; 20 trials with 8 pregnant women at 36 GWs in each trial.
Trial design D: Single oral dose of 150 mg caffeine [24]; 20 trials with 20 women in each trial and simulated at 0, 4, 8, 12, 16, 20, 24, 28, 32, 36, and 40 GWs.
Trial design E: Single oral dose of 177 mg caffeine [27]; 20 trials with 10 women in each trial and simulated at 0, 10–12, 15–18, 23–25, 31– 33, and 37–39 GWs.
Theophylline
Theophylline is a drug commonly used to treat asthma. It undergoes extensive hepatic metabolism (~ 90%), mainly by CYP1A2 (~ 80%) and CYP2E1 (~ 10%). The kidneys contribute by about 10% to the overall clearance of the drug [29,30,31].
Model building
The compound file “Theophylline” within the Simcyp Simulator was used keeping all the default parameters for metabolism. However, the distribution was described using a full-PBPK model where the tissue Kps were predicted using the Rodgers and Rowland method [22]. All Kps values were increased by 1.2 fold to achieve the observed volume of distribution at steady state. Absorption parameters were predicted within the Simulator using theophylline lipophilicity (LogPow) within the mechanistic permeability (MechPeff) model. Adequacy of these settings for the non-pregnant group were checked against observed data in a group of asthmatic (male and female) patients [32] before applying the model to the virtual pregnant population.
PK during pregnancy
Theophylline pharmacokinetics were evaluated throughout the pregnancy period at different gestational weeks in asthmatic, non-smoker, women after oral administration [33]. The following trial designs were set for model building and prediction during pregnancy to match the clinical studies:
Trial design A: Single oral dose of 7.3 mg/kg theophylline [32]; 20 trials with 10 subjects (5 female) in each trial aged 24–47 years.
Trial design B: Single oral dose of 7.6 mg/kg theophylline [32]; 20 trials with 10 subjects (5 female) in each trial aged 22–57 years.
Trial design C: Multiple oral dose regimen of 259 mg theophylline at 12 h interval for 4 days, followed by 259 mg oral theophylline on Day 5 [33]; 20 trials with 10 non-pregnant women in each trial.
Trial design D: Multiple oral dose regimen of 259 mg theophylline at 12 h interval for 4 days, followed by 259 mg oral theophylline on Day 5 [33]; 20 trials of 10 pregnant women (13–19 GWs).
Trial design E: Multiple oral dose regimen of 259 mg theophylline at 12 h interval for 4 days, followed by 259 mg oral theophylline on Day 5 [33]; 20 trials of 10 pregnant women (23–28 GWs).
Trial design F: Multiple oral dose regimen of 259 mg theophylline at 12 h interval for 4 days, followed by 259 mg oral theophylline on Day 5 [33]; 20 trials of 10 pregnant women (34–39 GWs).
Metoprolol
Metoprolol is eliminated by CYP2D6, CYP3A4 and excreted in urine. The average contribution of these routes in extensive metabolizers (EMs) are 80%, 9% and 11%, respectively. Its hepatic extraction ratio (EH) ranged from 0.35 to 0.77 for EMs and from 0.15 to 0.25 for poor metabolizers (PMs) [34].
Model building
The metoprolol compound file within the Simulator was selected with the default settings, but instead of the minimal PBPK model the full PBPK distribution model was used according to the Rodgers and Rowland method [22] without any adjustment to the calculated tissue Kps. The absorption profile of the model was described by the Advanced Dissolution, Absorption and Metabolism (ADAM) model, which accounts for physiological parameters relevant to the absorption phase of the drug [35]. The immediate release solid formulation option was used and the intrinsic solubility input for metoprolol was predicted using built-in calculator within the Simulator. These settings were made to establish a baseline model for the non-pregnant women after intravenous and oral administration of metoprolol [19].
PK during pregnancy
Metoprolol pharmacokinetics were investigated in few clinical studies during pregnancy. In one study, 5 hypertensive pregnant women at 35–38 GWs received a single intravenous dose of 10 mg metoprolol and 3 days later they received a single oral dose of 100 mg metoprolol. These doses were repeated 3–6 months after delivery to the same women [19]. It was concluded that the greater metoprolol clearance during pregnancy resulted from increased hepatic metabolism of the drug.
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after administrations of metoprolol:
Trial design A: A single intravenous dose of 10 mg metoprolol [19]; 20 trials of 5 non-pregnant women aged 20–36 years.
Trial design B: A single oral dose of 100 mg metoprolol [19, 36]; 20 trials of 13 non-pregnant women aged 20–36 years.
Trial design C: A single oral dose of 100 mg metoprolol [37]; 20 trials of 16 non-pregnant women aged 18–40 years, who were EMs (n = 16) with respect to CYP2D6.
Trial design D: A single oral dose of 100 mg metoprolol [37]; 20 trials of 4 non-pregnant women aged 18–40 years, who were PMs (n = 4) with respect to CYP2D6.
Trial design E: A single intravenous dose of 10 mg metoprolol [19]; 20 trials of 5 pregnant women aged 20–36 years at 35–38 GWs.
Trial design F: A single oral dose of 100 mg metoprolol [19, 36]; 20 trials of 13 pregnant women aged 20–36 years at 35–38 GWs.
Trial design G: A single oral dose of 100 mg metoprolol to 13 pregnant women, who were EMs with respect to CYP2D6 [38]; 20 trials of 13 pregnant women aged 26–33 years at 39 GWs.
Trial design H: Multiple oral dose of 50 mg metoprolol twice daily [39]; 20 trials of 6 pregnant women aged 27–37 years at 40 GWs.
Propranolol
Propranolol is an antihypertensive drug with high hepatic first pass elimination mainly via metabolic clearance. The fractional contribution of the metabolic pathways in the healthy non-pregnant population is about 0.21, 0.59, 0.17, and 0.024 for CYP1A2, CYP2D6, glucuronidation, and CYP2C19, respectively, while only 0.4% of the dose is eliminated by the kidney (see [40]).
Model building
A compound file for propranolol in healthy population has been published, where the drug elimination was described by enzyme kinetics, mainly, by CYP1A2, CYP2D6 and glucuronidation with a minor contribution from CYP2C19 [40]. Gender-differences in propranolol metabolism have been shown [41, 42] and hence systemic clearance data in non-pregnant women [43] was used for assigning intrinsic clearance for different metabolizing enzymes based on the fractional contribution from these enzymes. The disposition kinetics were described using the full PBPK distribution model. The tissues to plasma partition coefficients predicted within the Simulator using the Rodgers and Rowland method [22] to recover an intravenous profile reported for non-pregnant women [43] without any additional adjustment. The ADAM model was used to predict the absorption parameters for oral dosing in non-pregnant women [43]. Other physicochemical input parameters were taken from the published file [40].
PK during pregnancy
Propranolol pharmacokinetics during pregnancy were studied after propranolol was administered to pregnant women between 32 and 36 GWs and when at least 6 weeks postpartum in the same subjects. On both occasions, women were administered propranolol 120 mg orally or 10 mg intravenously in randomized order with a minimum washout period of 1 week [43]. Another study evaluate propranolol pharmacokinetics in pregnant women (15–39 GWs) and compared the results to a non-pregnant women group after multiple doses of 50 mg propranolol administered orally [44].
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after an intravenous or an oral administration of propranolol:
Trial design A: Single intravenous dose of 10 mg propranolol [43]; 20 trials of 5 healthy non-pregnant women aged 20–45 years.
Trial design B: Single intravenous dose of 10 mg propranolol [43]; 20 trials of 5 healthy pregnant women between 32 and 36 GWs aged 20–45 years.
Trial design C: Single oral dose of 120 mg propranolol [43]; 20 trials of 6 healthy non-pregnant women aged 20–45 years.
Trial design D: Single oral dose of 120 mg propranolol [43]; 20 trials of 6 healthy pregnant women between 32 and 36 GWs aged 20–45 years.
Trial design E: Multiple oral dose of 80 mg propranolol [43]; 20 trials of 9 healthy non-pregnant women aged 20–45 years.
Trial design F: Multiple oral dose of 80 mg propranolol [43]; 20 trials of 7 healthy pregnant women between 15 and 39 GWs aged 20–45 years.
Paroxetine
Paroxetine is a widely used antidepressant. It is completely absorbed after oral dosing and metabolized mainly by CYP2D6 in EMs (> 90%) [45]. The drug however is a potent inhibitor of CYP2D6.
Model building
The paroxetine compound file is selected from the Simulator compounds library. Default settings were kept, except the full PBPK distribution option was selected to represent different body tissues with the tissue Kps being predicted within the Simulator and accounting for membrane ion permeability [46]. Verification of this selection were checked against data from healthy non-pregnant subjects [45].
PK during pregnancy
Paroxetine kinetics in pregnancy has previously been explored in three studies [47,48,49] suggested that paroxetine plasma level is reduced during pregnancy compared with non-pregnant level, especially during the third trimester [48, 49].
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after an oral administration:
Trial design A: A multiple oral dose of 30 mg paroxetine every day for 14 days [45]; 20 trials of 9 healthy male subjects aged 20–30 years, who are CYP2D6 EMs.
Trial design B: A multiple oral dose of 30 mg paroxetine every day for 14 days [45]; 20 trials of 8 healthy male subjects aged 20–30 years, who are CYP2D6 PMs.
Trial design C: A multiple oral dose of 20 mg paroxetine every day [48]; 10 trials of 10 CYP2D6 EMs women aged 20–45 years over the whole period of pregnancy, from 0 to 40 GWs. Physiological parameters of the virtual pregnant women were updated on a daily basis within the simulation to account for the impact of pregnancy on the drug kinetics with time, i.e., time-varying covariates.
Trial design D: A multiple oral dose of 20 mg paroxetine every day; 10 trials of 10 women aged 20–45 years over the whole period of pregnancy, from 0 to 40 GWs. Default Caucasian CYP2D6 phenotype frequency were used. Physiological parameters of the virtual pregnant women were updated on a daily basis within the simulation to account for time-varying covariates.
Midazolam
Model building
The midazolam compound file within the Simulator was used. The distribution model was described by the full PBPK option where the predicted tissue Kps were reduced by half to match the iv data from non-pregnant women [50]. The first order oral absorption rate and fraction absorbed were predicted within the Simulator, using the default Caco-2 permeability data. A lag time of 0.2 h was introduced to recover the non-pregnant women concentration profile after an oral administration of midazolam [14]. These settings were retained to predict midazolam PK in pregnant women after intravenous and oral administration.
PK during pregnancy
Midazolam PK during pregnancy was studied between 28 GWs and term. Results from one study showed that midazolam exposure reduced by ~ 50% in 13 healthy pregnant women at 28–32 GWs compared to the exposure at 6–10 weeks post-partum [14]. Another study investigated midazolam kinetics after a single oral dose of 15 mg at term [15].
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after an intravenous or an oral administration:
Trial design A: Single intravenous dose of 0.2 mg/kg midazolam injected over 30 s [50]; 20 trials of 11 non-pregnant women aged 22–30 years.
Trial design B: Single oral dose of 2 mg midazolam [14]; 20 trials of 13 non-pregnant women aged 22–40 years.
Trial design C: Single oral dose of 2 mg midazolam [14]; 20 trials of 13 pregnant women at 28–32 GWs aged 22–40 years.
Trial design D: Single oral dose of 15 mg midazolam [15]; 20 trials of 8 pregnant women, at 40 GWs aged 40 years.
Trial design E: Single intravenous dose of 0.075 mg/kg midazolam given in conjunction with an elective caesarean section [15]; 20 trials of 8 women aged 20–32 years. For this simulation, the feto-placental unit volume and blood flow were re-set to zero to mimic the actual situation as the drug was given straight after delivery. CYP3A4 activity at 40 GWs was assumed in this case.
Nifedipine
Model Building
Nifedipine is an antihypertensive drug that is mainly metabolized by CYP3A4. The drug is completely absorbed from the gut, but due to its pre-systemic metabolism its bioavailability is about 56% to 77% [51]. The drug is available as immediate and controlled release formulations. The nifedipine compound file in the Simulator compounds library was used. Default settings were kept, except the absorption was described using the ADAM model to allow formulation specific predictions (see virtual trial design settings below). Other default nifedipine file settings used include the full PBPK distribution model that accounts for membrane ion permeability [46] and metabolism enzyme kinetics (mainly CYP3A4).
PK during pregnancy
Nifedipine kinetics during pregnancy were evaluated in different studies between 24 and 36 GWs [52], 26–35 GWs [53] and between 34 and 40 GWs [54].
The following trial designs were set for model building and prediction during pregnancy to match the clinical studies after an oral administration:
Trial design A: Single oral dose of 10 mg nifedipine immediate release capsule [55]; 20 trials of 7 (3 female) healthy non-pregnant subjects aged between 21 and 23 years.
Trial design B: Single oral dose of 10 mg nifedipine immediate release capsule [56]; 20 trials of 12 healthy male subjects aged between 21 and 28 years.
Trial design C: Single oral dose of 20 mg immediate release nifedipine [57]; 20 trials of 6 (2 female) healthy non-pregnant subjects aged between 20 and 30 years.
Trial design D: Multiple oral dose of 20 mg immediate release nifedipine [52]; 20 trials of 20 pregnant women aged between 20 to 40 years at 24–36 GWs.
Trial design E: Multiple oral doses of 10 mg nifedipine immediate release formulation administered every 6 h for total of 9 doses (54 h study duration) [53]; 20 trials of 15 pregnant women aged between 20 to 34 years at 26–35 GWs.
Trial design F: Multiple oral doses of 20 mg nifedipine controlled release formulation administered every 12 h for 3 days [54]; 20 trials of 12 pregnant women aged between 20–40 years with their gestational age ranging between 34 and 40 weeks. Similar to the clinical study, PK evaluations were evaluated on the last day on fasted state. An in vitro dissolution profile [58] was incorporated in this simulation to describe absorption for the slow release formulation.
Trial design G: Single oral dose of 10 mg nifedipine [59]; 20 trials of 8 women aged between 20 to 35 years during the immediate postpartum period. For this simulation, the feto-placental unit volume and blood flow were re-set to zero to mimic the actual situation. CYP3A4 activity at 40 GWs was assumed.
Rilpivirine
Rilpivirine is an antiretroviral drug used in combination with other antiretroviral agents, to treat human immunodeficiency virus infection (HIV) and to reduce perinatal HIV transmission to the fetus in Pregnant Women with HIV Infection. Rilpivirine is prescribed in a standard dosage of 25 mg once a day with food to increase its bioavailability. The absolute bioavailability of rilpivirine is unknown due to lack of an intravenous formulation [60]. Rilpivirine is approximately 99.7% bound to plasma proteins (mainly albumin) and reaches its maximum concentration within 4–5 h. It undergoes oxidative metabolism in the liver that is mainly mediated by CYP3A4 [60] and to a lesser extent by other poorly characterized kinetics, thought to be mediated by CYP2C19, UGT1A1 and UGT1A4 [61]. The elimination half-life of rilpivirine is approximately 50 h. About 25% of the administered dose is recovered as unchanged rilpivirine in feces, while only trace amounts of unchanged rilpivirine (< 1% of dose) were detected in urine [60].
Model building
A published PBPK rilpivirine file was used with default elimination kinetics (approximately 75% of metabolism by CYP3A4, and 25% through other metabolic routes) [60]. The tissues to plasma partition coefficients were predicted using the Rodgers and Rowland method [22] in the Simulator without any adjustment to the calculated tissue Kps. Absorption was described by a lag time of 0.5 h and a first order rate constant of 0.1 h−1 to recover the observed concentration time profile in healthy subjects in the fed state [62]. The fraction absorbed was assumed to be 75% as 25% of the unchanged drug is recovered in feces. These modifications were verified using an independent dataset [63].
PK during pregnancy
Rilpivirine pharmacokinetics have been evaluated during the second and third trimesters showing highly variable kinetics. Its exposure decreases during second and third trimesters compared with postpartum [64,65,66]. These PK data are in HIV patients, who are usually anaemic and have lower haematocrit and albumin level. Therefore, the default physiological parameters within the Simulator for non-pregnant women were accordingly adjusted to a mean value of 28.3% haematocrit [67] and a mean value of 41.14 g/L for serum albumin [68]. The default variability for both parameters within the Simulator were retained.
The following trial designs were set for model building and prediction during pregnancy to match the design of clinical studies after an oral administration of rilpivirine:
Trial design A: Single oral dose of 25 mg rilpivirine [62]; 20 trials of 113 healthy non-pregnant subject (31% female) aged 18–55 years.
Trial design B: Single oral dose of 75 mg rilpivirine [63]; 20 trials of 20 healthy non-pregnant subjects (10% female) aged 21–55 years.
Trial design C: Multiple oral dose of 25 mg rilpivirine for 14 days [64]; 20 trials of 15 non-pregnant women aged 20–36 years.
Trial design D: Multiple oral dose of 25 mg rilpivirine for 14 days [65]; 20 trials of 11 non-pregnant women aged 21–36 years.
Trial design E: Multiple oral dose of 25 mg rilpivirine for 14 days [66]; 20 trials of 28 non-pregnant women aged 20–45 years.
Trial design F: Multiple oral dose of 25 mg rilpivirine for 14 days [65]; 20 Trials of 15 pregnant subjects aged 21–36 years at 24–28 GWs.
Trial design G: Multiple oral dose of 25 mg rilpivirine for 14 days [66]; 20 Trials of 18 pregnant subjects aged 21–36 years at 24–28 GWs.
Trial design H: Multiple oral dose of 25 mg rilpivirine for 14 days [64]; 20 trials of 16 pregnant women aged 20–36 years at 30–37 GWs.
Trial design I: Multiple oral dose of 25 mg rilpivirine for 14 days [65]; 20 Trials of 13 pregnant subjects aged 21–36 years at 34–38 GWs.
Trial design J: Multiple oral dose of 25 mg rilpivirine for 14 days [66]; 20 Trials of 30 pregnant subjects aged 21–36 years at 31–40 GWs.
Results
Within the Simulator, the activity of CYP1A2, CYP2D6 and CYP3A4 during pregnancy were determined by the absolute abundance value of the enzyme in each non-pregnant virtual subject. The generated net expression of these enzymes in the liver during pregnancy are shown in Fig. 2. The following explains the simulation results for each investigated drug.

Activity and expression of CYP1A2, CYP2D6 and CYP3A4 enzymes during pregnancy. a relative activity to non-pregnant women. b, c and d absolute liver abundances (solid lines indicate population means for tracked 2000 virtual pregnant women; shaded areas indicate the 5th and 95th predicted percentiles)
Caffeine
The predicted caffeine concentration profiles for non-pregnant women are given in Fig. 3a and b. Model prediction for caffeine concentration time profile in pregnant women are shown in Fig. 3c–f. Predictions in all these scenarios are in good agreement with the clinical observations (Fig. 3). These plots also demonstrated that the pregnancy PBPK model replicated the observed higher maternal plasma exposure at term in pregnant women compared with non-pregnant women. The predicted continuous rise in the caffeine half-lives during pregnancy are in line with the observed data (Fig. 3d and e). A comparison of observed and predicted PK parameters for caffeine are given in Table 1.

Predicted (lines) vs observed (circles) caffeine PK in non-pregnant and pregnant women over time. Continuous bold lines are the predicted population means, grey lines are the means of each sub-trial, broken lines are the predictive 5th and 95th percentiles, and the error bars represent the standard deviations for the observed data. a Systemic caffeine plasma concentration in the 12 non-pregnant women (observed data from [21]). b Mean caffeine level in 4 non-pregnant pregnant women (observed data from [23]). c Mean caffeine PK profile in 8 pregnant women at 36 GWs (observed data from [23]). d Predicted and observed caffeine half-lives at different gestational weeks (means of observed data from closed circles [24], open circles [27]). e Predicted and observed mean caffeine (oral) clearance at different gestational weeks (means of observed data from [27]). f Predicted and observed caffeine elimination rate at different GWs (observed data from [27]). Solid lines and shaded area in d, e and f represent the predicted population means and 5th–95th percentiles for the generated virtual pregnant population at different gestational weeks. See “Methods” section for trial settings
Theophylline
Theophylline simulations for the baseline model in non-pregnant women are shown in Fig. 4a. Simulations were in agreement with the observed individual data after oral administration. The PBPK model predictions during pregnancy (Fig. 4b, c and d) described the observed individual profiles adequately at different gestational weeks. Comparison of the predicted PK parameters obtained for the simulated profiles with those available from clinical study are given in Fig. 4e and f.

Predicted (lines) vs observed (circles) theophylline pharmacokinetics. Plasma concentration time profiles in non-pregnant subjects (a and b [32]) non-pregnant women (c [33]) and pregnant women at 13–19 GWs (d [33]), 23–28 GWs (e [33]), and 34–39 GWs (f [33]). Continuous bold lines are the predicted population means, grey lines are the means of each sub-trial, and broken lines are the predicted 5th and 95th percentiles. See “Methods” section for trial settings. PK parameters at different gestational weeks (g Half-life [33]; h Clearance (oral) [33]). Shaded area are the predicted 5th and 95th percentiles with the predicted population mean shown as a solid line. Circles are observed individual data
Metoprolol
Predicted systemic concentration profiles of metoprolol in plasma as well as derived PK parameters were generally in agreement with the observed data. Metoprolol PBPK model predictions for plasma level in non-pregnant women after intravenous and oral administration are given in Fig. 5a–d. Predicted metoprolol concentration–time profiles in pregnancy compared to the clinical data are shown in Figs. 5e, f, g, and h. Calculated pharmacokinetic parameters of metoprolol from simulated profiles are given in Table 2, in comparison with those obtained for clinical profiles.

Predicted (lines) vs observed (circles) metoprolol plasma concentration in non-pregnant (a–d) and pregnant (e–h) women. Observed data in a, b, e, f and h are individual data, while in c, d and g are mean data. Bold continuous lines are the predicted means, grey thin lines are the trial means and the broken lines are the 5th and 95th percentiles. Plots a, b, c and d are profiles in non-pregnant women after either single intravenous (a [19]) or single oral (b [19] closed circles & [36] open circles), (c EMs [37]) and (d PMs [37]) administration. Plots e, f, g, and h are profiles in pregnant women after either single intravenous (e [19]), single oral (f [19] closed circles) & ( [36] open circles), and g (EMs [38])) or multiple oral (h [39]) administration. See “Methods” section for trial settings
Propranolol
Propranolol simulations for the baseline model in non-pregnant women are shown in Fig. 6a and b for the model building step. Simulations were in agreement with the observed data after both intravenous and oral administration. The pregnancy PBPK model slightly under-predicted the mean observed intravenous profile in four subjects. However, the observed data are still within the predicted range (Fig. 6c). On the other hand, the observed profiles after single and oral administration (Fig. 6d and f) were adequately predicted using the pregnancy PBPK model. Comparison of the predicted PK parameters obtained for the simulated profiles with those available from clinical study are given in Table 3.

Propranolol systemic concentration in plasma in non-pregnant (a, b and e) and pregnant women. Continuous bold lines are predicted population means, each grey line represents the predicted mean of each simulated sub-trial, broken lines are predictive 5th and 95th percentiles, and error bars represent standard deviations for the observed data (circles). (a: single 10 mg iv infusion to 5 non-pregnant women [43]; b single 120 mg oral to 6 non-pregnant women [43]; c single 10 mg iv infusion to 5 pregnant women at 32–36 GWs [43]; d single 120 mg oral to 6 pregnant women at 32–36 GWs [43]; e multiple 80 mg oral to 9 non-pregnant women [44]; f multiple 80 mg oral to 7 pregnant women at 15–39 GWs [44]. See “Methods” section for trial settings
Paroxetine
Paroxetine PBPK predictions for the non-pregnant populations classified as CYP2D6 EMs or PMs are in agreement with the observed profiles after single and multiple doses (Fig. 7a and b). The pregnancy PBPK also showed that mean simulated concentration–time profiles in the EMs (Fig. 7c) and general (Fig. 7d) populations are closely following the observed profiles after multiple‐dose administration. In addition, the 95% confidence intervals generally contained most of the observed data points.

Predicted (lines) vs observed (circles) paroxetine plasma concentrations. Plot a Healthy male subjects, who were EMs [45] and Plot B PMs [45] to CYP2D6. Plot c Pregnant women, who were EMs [48] and general Caucasian population phenotypes (red circles from [49]; green circles [74]). Note the x-axis is in days for plots c and d. Continuous bold lines are predicted population means, each grey line represents the predicted mean of each simulated sub-trial, and broken lines are predictive 5th and 95th percentiles. See “Methods” section for trial settings
Midazolam
The midazolam simulation results show good agreement between the predicted and observed drug level in the systemic circulation in both non-pregnant and pregnant women (Fig. 8). Comparison of the PK parameters obtained for the simulated profiles with those from clinical studies (Table 4) shows also good agreement and all PK parameters were within 2-fold of the observed values.

Predicted (lines) vs observed (circles) systemic midazolam plasma concentration profiles in non-pregnant and pregnant women. Continuous bold lines are the predicted population means, each grey line represents the predicted mean of each simulated sub-trial, broken lines are the predicted 5th and 95th percentiles, and error bars represent standard deviations for the observed data. Plots a and b are for non-pregnant women after a intravenous (a mean profile [50]) and oral (individual profiles (see [52])) administration. Plots c and d are for pregnant women after oral administration at 28–32GWs (see [52]) and at term [15]. Plot e is after intravenous administration given in conjunction with an elective caesarean section [15]. See “Methods” section for each scenario settings
Nifedipine
The predicted nifedipine plasma concentration profiles in healthy male, non-pregnant and pregnant women for IR and CR formulations were in agreement with the clinical observations (Fig. 9). Observed concentrations were within the predicted 5th and 95th percentiles. Predicted vs observed ratios for various pharmacokinetics parameters are given in Table 5, all the predictions were within 2-fold of the observed values.

Predicted (lines) vs observed (circles) systemic nifedipine plasma concentration profiles in non-pregnant and pregnant women. Continuous bold lines are the predicted population means, each grey line represents the predicted mean of each simulated sub-trial, broken lines are the predicted 5th and 95th percentiles, and error bars represent standard deviations for the observed data. The nifedipine level in non-pregnant women after oral administration are given in plots a [55], b [56] and c [57]. The nifedipine level in pregnant women after oral administration are given in plots d (24–36 GWs [52]), e (26–35 GWs [53] and f (34.4–39.6 GWs [54]). Plot g is the observed plasma level of nifedipine for a single oral dose given immediately after birth [59]. See “Methods” section for trial settings
Rilpivirine
Rilpivirine predictions for the baseline model in healthy subjects are shown in Fig. 10a and b for verifying the developed PBPK model. Predicted plasma rilpivirine concentration profiles in non-pregnant HIV women are shown in Fig. 10c–e. Predictions were in good agreement with the observed data, except for Tran’s study [66] in non-pregnant women (Fig. 10e). However, the observed data are still within the predicted range. Predicted profiles during second (Fig. 10f and j) and third (Fig. 10h, i and j) trimesters were all in good agreement with the observed profiles. Comparison of the predicted PK parameters obtained for the simulated profiles with those available from clinical study are given in Table 6.

Rilpivirine systemic concentration in plasma after single dose in healthy subjects (a [62] and b [63]) and at steady state in non-pregnant HIV women (c [64], d [65] and e [66]), pregnant women at second trimester (f [65] and g [66]) and third trimester (h [64], i [65], j [66]). Continuous bold lines are predicted population means, each grey line represents the predicted mean of each simulated sub-trial, broken lines are predictive 5th and 95th percentiles, and error bars represent standard deviations for the observed data (circles). Data in pregnant women are at 24–28 GWs (f [65]), 20–28 GWs (g [66]), 30–37 GWs (h [64]), 34–38 GWs (i [65]), and 31–40 GWs (j [66]). See “Methods” section for trial settings
Discussion
The current study utilizes the PBPK approach to assess, longitudinally, the impact of temporal changes to CYP1A2, CYP2D6 and CYP3A4 metabolizing enzymes on drug’s disposition during pregnancy using probe substrates for each enzyme at different gestational weeks. The predictions were compared against data taken from independent studies. A quantitative longitudinal description of these changes are essential for dose adjustment during pregnancy. The activity of these enzymes are part of the pregnancy population. Drugs that are metabolized by more than one of these enzymes will be affected by these time-varying equations. For example, propranolol is metabolized by CYP2D6 and CYP1A2, so its clearance is affected by the activity of the two enzymes at the same time at any gestational age. This is also the case for metoprolol, which is metabolized by CYP2D6 and CYP3A4. Other PBPK model parameters that account for gestational age dependent physiology and inter-individual variability [1] were also incorporated within the model to prediction of drugs kinetics at different gestational weeks. The approach used for integrating pregnancy-related physiological parameters together with compound dependent parameters within the PBPK model to predict the maternal concentration time profiles has been previously described in detail [16].
The reduction of CYP1A2 activity during pregnancy adequately describes the changes in caffeine pharmacokinetics during the whole gestational period reported in different studies [23, 24, 27]. Likewise, predicted theophylline plasma profiles and PK parameters were in good agreement with the observed data at different gestational ages.
The increase in CYP2D6 activity, as investigated using the metoprolol clearance from CYP2D6 EMs, during pregnancy adequately describes the changes in metoprolol kinetics reported in different studies indicating the suitability of this function to reflect in vivo changes of CYP2D6 activity during pregnancy. Due to using general population phenotypes and simulating profiles for a range of gestational weeks (35–38 GWs), a large variability was observed in the predicted profiles, which is also comparable to the observed variability in pregnant individuals [19, 36]. There was an over-prediction of metoprolol concentration in Lindeberg’s study [39] (Fig. 5h), however individuals were under co-medications with multiple drugs and no information about their phenotypes was provided.
Propranolol is also considered to be a CYP2D6 substrate, which undergoes significant first pass metabolism by the liver. Its pharmacokinetic parameters are highly variable between subjects (~ 100% CV). The observed fold increase in clearance during third trimester compared to non-pregnant women was 1.2 (predicted 1.3) and 1.5 (predicted 2.5) fold after intravenous and oral administration, respectively (Table 3). Predicted profiles were adequately described the observed data. The predicted mean after an oral dose in non-pregnant women slightly over-predicted the observed mean in 6 subjects (Fig. 6b), but under-predicted the observed mean concentration profile in another study after multiple doses (Fig. 6e). During pregnancy, clinical studies indicated no significant differences in propranolol kinetics across the three trimesters vs the postnatal period [43, 44, 69]. This is probably due to the following reasons. First, two counteracting enzymes affecting the clearance, i.e., the continuous increase in CYP2D6 activity is partially cancelled by the continuous decrease in CYP1A2 activity over the gestational time. Second, propranolol plasma concentrations were not different in subjects with extensive vs poor CYP2D6 phenotypes [70]. Propranolol and/or any of its metabolites may exert an inhibitory effect on one or more metabolic pathways of propranolol itself. Finally, propranolol metabolism is both regio- and stereo-selective [71, 72]. It should be pointed out that the racemic propranolol, rather than it enantiomers, was considered in the developed model.
Paroxetine is another CYP2D6 probe substrate, however it is also a potent CYP3A4 and CYP2D6 inhibitor [73], which impairs its own clearance [45]. Paroxetine auto-inhibition mechanisms were incorporated in the model. Despite these observations, simulations were performed for these compounds. Results of these predictions were in agreement with the clinically observed kinetics in pregnancy, which reinforces the utility of the pregnancy PBPK model.
The applied quantitative description of the increase in CYP3A4 activity during pregnancy resulted in good recovery of observed midazolam exposure during pregnancy. Similarly, good recovery of observed nifedipine exposure from different independent studies was obtained when the gestational impact on CYP3A4 activity was accounted for in the model. There was significant inter-subject variability in the observed data for both IR and CR nifedipine formulation studies, especially around Cmax. Currently, there are limited and conflicting data on gastrointestinal changes during pregnancy that can affect drug absorption from the gastrointestinal tract [1], therefore in the ADAM model the physiological parameters of non-pregnant women were used. The quantified increases in CYP3A4 activity during pregnancy resulted in good description of the observed reduction of rilpivirine exposure during second and third trimesters compared with postpartum level. The predicted mean PK parameters were also within 1.5-fold of the observed values. There is an over-prediction of the observed profiles in Tran’s study for postpartum, however this trend was not observed for postpartum women in other studies. HIV patients are usually under co-medications as it is the case for all rilpivirine studies, which can affect the drug exposure and might be the reason for the observed discrepancy. In the current analysis, co-medication was not taken into account during PK predictions.
For some drugs, like caffeine the absorption is fast and complete as its permeability through the gut wall is high and it does not undergo gut metabolism due to the absence of CYP1A2 in the gut, hence the default first order absorption model is used. For rilpivirine, the absorption is poorly understood and cannot be deconvoluted due to the absence of intravenous formulation and as a result the first order absorption model is used. For other compounds where the default absorption model is ADAM or information on formulation are available, the ADAM model was used. Since this research work mainly focuses on the disposition kinetics, changes to the absorption settings were kept minimal. Mechanistically, the ADAM model is preferred over the first order model and this could be an enhancement to the rilpivirine file in the future.
Due to the nature of science evolution, reported PK results from historical and recent clinical studies were presented in different ways, for example earlier studies were not designed to evaluate the impact of phenotypes of CYP2D6. Historically, different assays were used to quantify the drug concentration with different sensitivity levels. Few studies reported lumped PK parameters derived from wide gestational age range additionally they did not provide individual concentration time profiles or distribution of the individuals’ gestational age within the studied gestational week ranges. Distribution of the observed PK parameters were reported in some cases as ranges, while others reported only standard error of the mean, which further challenged comparing results between studies. There is also information missing in the original studies regarding the trial setting used and the inclusion criteria. This resulted in replicating and comparing studies separately. Absence of cross-over type studies, where individuals are their own control, and as in the case of nifedipine, the complete absence of a reference non-pregnant women population further challenged the interpretation of the results. In most cases concentration profiles were read from logarithm plots. Further verification of these enzymes with compounds of diverse properties are required to build confidence in the model. Expanding the scope of the pregnancy PBPK model applications to include additional enzymes and transporters is of interest to obtain better insights in effects of gestation on the drug disposition during pregnancy.
The results of this study show the ability of the model to predict the exposure of drugs for the investigated enzymes. Therefore, it may be used to inform dosing during pregnancy for drugs where such information is not available. CYP1A2 enzyme is involved in the metabolism of some drugs. Its reduced activity during pregnancy can increase the systemic exposure of CYP1A2 substrates above the therapeutic window leading to toxicity and adverse effects on the developing fetus. On the other hand, the activity of both CYP3A4 and CYP2D6 increases during pregnancy, which can result in drug exposure below the effective concentration and consequently treatment failure. Furthermore, toxicity can occur indirectly due to an enhanced generation of toxic metabolite in the case of CYP3A4 and CYP2D6 enzymes. In all cases, the teratogenic effect can also occur at the recommended therapeutic dose and therefore dose modification should be carefully assessed.
Conclusion
Quantified changes in CYP1A2, CYP2D6 and CYP3A4 activity resulted in a good recovery of the observed profiles and PK parameters for their sensitive probes. Results obtained in this study demonstrate the utility of pregnancy PBPK modelling as a generic tool for predicting dosage requirements in pregnant patients with respect to substrates of specific enzymes. The pregnancy PBPK model should be viewed as a “live” model which undergoes continuous enhancements as knowledge of physiological and biochemical changes during pregnancy increases to expand the applications to other CYPs and non-CYPs enzymes as well as transporters. The level of confidence in these models at any given time reflects the state of existing knowledge and the models capability to incorporate such knowledge.
Abbreviations
- PBPK:
-
Physiologically-based pharmacokinetics
- GWs:
-
Gestational weeks
References
Abduljalil K, Furness P, Johnson TN, Rostami-Hodjegan A, Soltani H (2012) Anatomical, physiological and metabolic changes with gestational age during normal pregnancy: a database for parameters required in physiologically based pharmacokinetic modelling. Clin Pharmacokinet 51:365–396. https://doi.org/10.2165/11597440-000000000-00000
Abduljalil K, Badhan RKS (2020) Drug dosing during pregnancy-opportunities for physiologically based pharmacokinetic models. J Pharmacokinet Pharmacodyn E. https://doi.org/10.1007/s10928-020-09698-w
Wienkers LC, Heath TG (2005) Predicting in vivo drug interactions from in vitro drug discovery data. Nat Rev Drug Discov 4:825–833. https://doi.org/10.1038/nrd1851
Wilkinson GR (2005) Drug metabolism and variability among patients in drug response. N Engl J Med 352:2211–2221. https://doi.org/10.1056/NEJMra032424
Zanger UM, Schwab M (2013) Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 138:103–141. https://doi.org/10.1016/j.pharmthera.2012.12.007
Chen W, Xiao Y, Chen J, Liu J, Shao J, Li T, Zhu Y, Ma J, Gao Y, Wang J, Xu J, Lu Y, Jia L (2017) Sex-related pharmacokinetic differences and mechanisms of metapristone (RU486 metabolite). Sci Rep 7:17190. https://doi.org/10.1038/s41598-017-17225-0
Zucker I, Prendergast BJ (2020) Sex differences in pharmacokinetics predict adverse drug reactions in women. Biol Sex Differ 11:32. https://doi.org/10.1186/s13293-020-00308-5
Beierle I, Meibohm B, Derendorf H (1999) Gender differences in pharmacokinetics and pharmacodynamics. Int J Clin Pharmacol Ther 37:529–547
Tracy TS, Venkataramanan R, Glover DD, Caritis SN, National Institute for Child H, Human Development Network of Maternal-Fetal-Medicine U (2005) Temporal changes in drug metabolism (CYP1A2, CYP2D6 and CYP3A Activity) during pregnancy. Am J Obstet Gynecol 192:633–639. https://doi.org/10.1016/j.ajog.2004.08.030
Tsutsumi K, Kotegawa T, Matsuki S, Tanaka Y, Ishii Y, Kodama Y, Kuranari M, Miyakawa I, Nakano S (2001) The effect of pregnancy on cytochrome P4501A2, xanthine oxidase, and N-acetyltransferase activities in humans. Clin Pharmacol Ther 70:121–125. https://doi.org/10.1067/mcp.2001.116495
Ryu RJ, Eyal S, Easterling TR, Caritis SN, Venkataraman R, Hankins G, Rytting E, Thummel K, Kelly EJ, Risler L, Phillips B, Honaker MT, Shen DD, Hebert MF (2016) Pharmacokinetics of metoprolol during pregnancy and lactation. J Clin Pharmacol 56:581–589. https://doi.org/10.1002/jcph.631
Rostami-Hodjegan A, Kroemer HK, Tucker GT (1999) In-vivo indices of enzyme activity: the effect of renal impairment on the assessment of CYP2D6 activity. Pharmacogenetics 9:277–286. https://doi.org/10.1097/00008571-199906000-00002
Borges S, Li L, Hamman MA, Jones DR, Hall SD, Gorski JC (2005) Dextromethorphan to dextrorphan urinary metabolic ratio does not reflect dextromethorphan oral clearance. Drug Metab Dispos 33:1052–1055. https://doi.org/10.1124/dmd.104.003459
Hebert MF, Easterling TR, Kirby B, Carr DB, Buchanan ML, Rutherford T, Thummel KE, Fishbein DP, Unadkat JD (2008) Effects of pregnancy on CYP3A and P-glycoprotein activities as measured by disposition of midazolam and digoxin: a University of Washington specialized center of research study. Clin Pharmacol Ther 84:248–253. https://doi.org/10.1038/clpt.2008.1clpt20081
Kanto J, Sjovall S, Erkkola R, Himberg JJ, Kangas L (1983) Placental transfer and maternal midazolam kinetics. Clin Pharmacol Ther 33:786–791. https://doi.org/10.1038/clpt.1983.107
Gaohua L, Abduljalil K, Jamei M, Johnson TN, Rostami-Hodjegan A (2012) A pregnancy physiologically based pharmacokinetic (p-PBPK) model for disposition of drugs metabolized by CYP1A2, CYP2D6 and CYP3A4. Br J Clin Pharmacol 74:873–885. https://doi.org/10.1111/j.1365-2125.2012.04363.x
Abduljalil K, Jamei M, Rostami-Hodjegan A, Johnson TN (2014) Changes in individual drug-independent system parameters during virtual paediatric pharmacokinetic trials: introducing time-varying physiology into a paediatric PBPK model. AAPS J 16:568–576. https://doi.org/10.1208/s12248-014-9592-9
Abduljalil K, Pan X, Pansari A, Jamei M, Johnson TN (2020) Preterm physiologically based pharmacokinetic model. Part II: Applications of the model to predict drug pharmacokinetics in the preterm population. Clin Pharmacokinet 59:501–518. https://doi.org/10.1007/s40262-019-00827-4
Hogstedt S, Lindberg B, Peng DR, Regardh CG, Rane A (1985) Pregnancy-induced increase in metoprolol metabolism. Clin Pharmacol Ther 37:688–692
Greenblatt DJ, Abernethy DR, Locniskar A, Harmatz JS, Limjuco RA, Shader RI (1984) Effect of age, gender, and obesity on midazolam kinetics. Anesthesiology 61:27–35
Kim MK, Nightingale C, Nicolau D (2003) Influence of sex on the pharmacokinetic interaction of fleroxacin and ciprofloxacin with caffeine. Clin Pharmacokinet 42:985–996. https://doi.org/10.2165/00003088-200342110-00004
Rodgers T, Rowland M (2006) Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci 95:1238–1257. https://doi.org/10.1002/jps.20502
Brazier JL, Ritter J, Berland M, Khenfer D, Faucon G (1983) Pharmacokinetics of caffeine during and after pregnancy. Dev Pharmacol Ther 6:315–322
Knutti R, Rothweiler H, Schlatter C (1981) Effect of pregnancy on the pharmacokinetics of caffeine. Eur J Clin Pharmacol 21:121–126
Parsons WD, Pelletier JG (1982) Delayed elimination of caffeine by women in the last 2 weeks of pregnancy. Can Med Assoc J 127:377–380
Yu T, Campbell SC, Stockmann C, Tak C, Schoen K, Clark EA, Varner MW, Spigarelli MG, Sherwin CM (2016) Pregnancy-induced changes in the pharmacokinetics of caffeine and its metabolites. J Clin Pharmacol 56:590–596. https://doi.org/10.1002/jcph.632
Aldridge A, Bailey J, Neims AH (1981) The disposition of caffeine during and after pregnancy. Semin Perinatol 5:310–314
Haas DM, Hebert MF, Soldin OP, Flockhart DA, Madadi P, Nocon JJ, Chambers CD, Hankins GD, Clark S, Wisner KL, Li L, Renbarger JL, Learman LA (2009) Pharmacotherapy and pregnancy: highlights from the Second International Conference for Individualized Pharmacotherapy in Pregnancy. Clin Transl Sci 2:439–443. https://doi.org/10.1111/j.1752-8062.2009.00166.x
Rasmussen BB, Nielsen KK, Brosen K (1994) Determination of theophylline metabolites in human liver microsomes by high-performance liquid chromatography. Anal Biochem 222:9–13. https://doi.org/10.1006/abio.1994.1446
Cornish HH, Christman AA (1957) A study of the metabolism of theobromine, theophylline, and caffeine in man. J Biol Chem 228:315–323
Levy G, Koysooko R (1976) Renal clearance of theophylline in man. J Clin Pharmacol 16:329–332. https://doi.org/10.1002/j.1552-4604.1976.tb01528.x
Hendeles L, Weinberger M, Bighley L (1977) Absolute bioavailability of oral theophylline. Am J Hosp Pharm 34:525–527
Gardner MJ, Schatz M, Cousins L, Zeiger R, Middleton E, Jusko WJ (1987) Longitudinal effects of pregnancy on the pharmacokinetics of theophylline. Eur J Clin Pharmacol 32:289–295. https://doi.org/10.1007/BF00607577
Lennard MS, Tucker GT, Silas JH, Freestone S, Ramsay LE, Woods HF (1983) Differential stereoselective metabolism of metoprolol in extensive and poor debrisoquin metabolizers. Clin Pharmacol Ther 34:732–737. https://doi.org/10.1038/clpt.1983.242
Jamei M, Turner D, Yang J, Neuhoff S, Polak S, Rostami-Hodjegan A, Tucker G (2009) Population-based mechanistic prediction of oral drug absorption. AAPS J 11:225–237. https://doi.org/10.1208/s12248-009-9099-y
Hogstedt S, Rane A (1993) Plasma concentration-effect relationship of metoprolol during and after pregnancy. Eur J Clin Pharmacol 44:243–246. https://doi.org/10.1007/BF00271365
Sharma A, Pibarot P, Pilote S, Dumesnil JG, Arsenault M, Belanger PM, Meibohm B, Hamelin BA (2005) Modulation of metoprolol pharmacokinetics and hemodynamics by diphenhydramine coadministration during exercise testing in healthy premenopausal women. J Pharmacol Exp Ther 313:1172–1181. https://doi.org/10.1124/jpet.104.081109
Antunes Nde J, Cavalli RC, Marques MP, Moises EC, Lanchote VL (2015) Influence of gestational diabetes on the stereoselective pharmacokinetics and placental distribution of metoprolol and its metabolites in parturients. Br J Clin Pharmacol 79:605–616. https://doi.org/10.1111/bcp.12523
Lindeberg S, Lundborg P, Regardh CG, Sandstrom B (1987) Disposition of the adrenergic blocker metoprolol and its metabolite OH-metoprolol in maternal plasma, amniotic fluid and capillary blood of the neonate. Eur J Clin Pharmacol 33:363–368. https://doi.org/10.1007/BF00637631
Rose RH, Turner DB, Neuhoff S, Jamei M (2017) Incorporation of the time-varying postprandial increase in splanchnic blood flow into a pbpk model to predict the effect of food on the pharmacokinetics of orally administered high-extraction drugs. AAPS J 19:1205–1217. https://doi.org/10.1208/s12248-017-0099-z
Walle T, Walle K, Mathur RS, Palesch YY, Conradi EC (1994) Propranolol metabolism in normal subjects: association with sex steroid hormones. Clin Pharmacol Ther 56:127–132. https://doi.org/10.1038/clpt.1994.115
Walle T, Walle UK, Cowart TD, Conradi EC (1989) Pathway-selective sex differences in the metabolic clearance of propranolol in human subjects. Clin Pharmacol Ther 46:257–263. https://doi.org/10.1038/clpt.1989.136
O'Hare MF, Kinney CD, Murnaghan GA, McDevitt DG (1984) Pharmacokinetics of propranolol during pregnancy. Eur J Clin Pharmacol 27:583–587
Hoffmann-Traeger A, Peiker G, Glockner R, Wittmann I, Balogh A, Henschel L (1987) The influence of phenobarbital on the pharmacokinetics of propranolol in pregnancy. Biol Res Pregnancy Perinatol 8:57–59
Sindrup SH, Brosen K, Gram LF, Hallas J, Skjelbo E, Allen A, Allen GD, Cooper SM, Mellows G, Tasker TC et al (1992) The relationship between paroxetine and the sparteine oxidation polymorphism. Clin Pharmacol Ther 51:278–287. https://doi.org/10.1038/clpt.1992.23
Fisher C, Simeon S, Jamei M, Gardner I, Bois YF (2019) VIVD: Virtual in vitro distribution model for the mechanistic prediction of intracellular concentrations of chemicals in in vitro toxicity assays. Toxicol In Vitro 58:42–50. https://doi.org/10.1016/j.tiv.2018.12.017
Misri S, Kim J, Riggs KW, Kostaras X (2000) Paroxetine levels in postpartum depressed women, breast milk, and infant serum. J Clin Psychiatry 61:828–832. https://doi.org/10.4088/jcp.v61n1104
Ververs FF, Voorbij HA, Zwarts P, Belitser SV, Egberts TC, Visser GH, Schobben AF (2009) Effect of cytochrome P450 2D6 genotype on maternal paroxetine plasma concentrations during pregnancy. Clin Pharmacokinet 48:677–683. https://doi.org/10.2165/11318050-000000000-00000
Westin AA, Brekke M, Molden E, Skogvoll E, Spigset O (2017) Selective serotonin reuptake inhibitors and venlafaxine in pregnancy: changes in drug disposition. PLoS ONE 12:e0181082. https://doi.org/10.1371/journal.pone.0181082
Avram MJ, Fragen RJ, Caldwell NJ (1983) Midazolam kinetics in women of two age groups. Clin Pharmacol Ther 34:505–508. https://doi.org/10.1038/clpt.1983.205
Raemsch KD, Sommer J (1983) Pharmacokinetics and metabolism of nifedipine. Hypertension 5:18–24. https://doi.org/10.1161/01.hyp.5.4_pt_2.ii18
Quinney SK, Mohamed AN, Hebert MF, Haas DM, Clark S, Umans JG, Caritis SN, Li L (2012) A semi-mechanistic metabolism model of CYP3A substrates in pregnancy: predicting changes in midazolam and nifedipine pharmacokinetics. CPT Pharmacometrics Syst Pharmacol 1:e2. https://doi.org/10.1038/psp.2012.5
Prevost RR, Akl SA, Whybrew WD, Sibai BM (1992) Oral nifedipine pharmacokinetics in pregnancy-induced hypertension. Pharmacotherapy 12:174–177
Filgueira GC, Filgueira OA, Carvalho DM, Marques MP, Moises EC, Duarte G, Lanchote VL, Cavalli RC (2015) Analysis of nifedipine in human plasma and amniotic fluid by liquid chromatography-tandem mass spectrometry and its application to clinical pharmacokinetics in hypertensive pregnant women. J Chromatogr B 993–994:20–25. https://doi.org/10.1016/j.jchromb.2015.04.030S1570-0232(15)00242-1[pii]
Rashid TJ, Martin U, Clarke H, Waller DG, Renwick AG, George CF (1995) Factors affecting the absolute bioavailability of nifedipine. Br J Clin Pharmacol 40:51–58. https://doi.org/10.1111/j.1365-2125.1995.tb04534.x
Foster TS, Hamann SR, Richards VR, Bryant PJ, Graves DA, McAllister RG (1983) Nifedipine kinetics and bioavailability after single intravenous and oral doses in normal subjects. J Clin Pharmacol 23:161–170. https://doi.org/10.1002/j.1552-4604.1983.tb02720.x
Holtbecker N, Fromm MF, Kroemer HK, Ohnhaus EE, Heidemann H (1996) The nifedipine-rifampin interaction. Evidence for induction of gut wall metabolism. Drug Metab Dispos 24:1121–1123
Teder K, Pepeloshev A, Matto V, Meos A (2013) Pharmacopoieal quality of non-expired and expired nifedipine formulations from Estonian and Russian Federation medicinal products market. Acta Pol Pharm 70:539–546
Barton JR, Prevost RR, Wilson DA, Whybrew WD, Sibai BM (1991) Nifedipine pharmacokinetics and pharmacodynamics during the immediate postpartum period in patients with preeclampsia. Am J Obstet Gynecol 165:951–954. https://doi.org/10.1016/0002-9378(91)90446-x
U.S. Food and Drug Administration (FDA) (2011) New Drug Application 202022 (Rilpivirine): Clinical Pharmacology and Biopharmacetics Review(s). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202022Orig1s000ClinPharmR.pdf. Accessed 20 Jul 2020
Lade JM, Avery LB, Bumpus NN (2013) Human biotransformation of the nonnucleoside reverse transcriptase inhibitor rilpivirine and a cross-species metabolism comparison. Antimicrob Agents Chemother 57:5067–5079. https://doi.org/10.1128/AAC.01401-13
Mehta R, Wolstenholme A, Di Lullo K, Fu C, Joshi S, Crauwels H, givens N, vanveggel s, wynne b, adkison k (2018) bioequivalence of a fixed-dose combination tablet of the complete two-drug regimen of dolutegravir and rilpivirine for treatment of HIV-1 infection. Antimicrob Agents Chemother. https://doi.org/10.1128/AAC.00748-18
Crauwels HM, van Heeswijk RP, Buelens A, Stevens M, Boven K, Hoetelmans RM (2013) Impact of food and different meal types on the pharmacokinetics of rilpivirine. J Clin Pharmacol 53:834–840. https://doi.org/10.1002/jcph.107
Schalkwijk S, Colbers A, Konopnicki D, Gingelmaier A, Lambert J, van der Ende M, Molto J, Burger D, Pharmacokinetics of newly developed antiretroviral agents in HIVipwN (2017) Lowered rilpivirine exposure during the third trimester of pregnancy in human immunodeficiency virus type 1-infected women. Clin Infect Dis 65:1335–1341. https://doi.org/10.1093/cid/cix534
Osiyemi O, Yasin S, Zorrilla C, Bicer C, Hillewaert V, Brown K, Crauwels HM (2018) Pharmacokinetics, antiviral activity, and safety of rilpivirine in pregnant women with hiv-1 infection: results of a phase 3b, multicenter, open-label study. Infect Dis Ther 7:147–159. https://doi.org/10.1007/s40121-017-0184-8
Tran AH, Best BM, Stek A, Wang J, Capparelli EV, Burchett SK, Kreitchmann R, Rungruengthanakit K, George K, Cressey TR, Chakhtoura N, Smith E, Shapiro DE, Mirochnick M, Team IPP (2016) Pharmacokinetics of rilpivirine in HIV-infected pregnant women. J Acquir Immune Defic Syndr 72:289–296. https://doi.org/10.1097/QAI.0000000000000968
Obirikorang C, Yeboah FA (2009) Blood haemoglobin measurement as a predictive indicator for the progression of HIV/AIDS in resource-limited setting. J Biomed Sci 16:102. https://doi.org/10.1186/1423-0127-16-102
Shah S, Smith CJ, Lampe F, Youle M, Johnson MA, Phillips AN, Sabin CA (2007) Haemoglobin and albumin as markers of HIV disease progression in the highly active antiretroviral therapy era: relationships with gender. HIV Med 8:38–45. https://doi.org/10.1111/j.1468-1293.2007.00434.x
Smith MT, Livingstone I, Eadie MJ, Hooper WD, Triggs EJ (1983) Chronic propranolol administration during pregnancy. Matern Pharm 25:481–490
Lennard MS, Jackson PR, Freestone S, Tucker GT, Ramsay LE, Woods HF (1984) The relationship between debrisoquine oxidation phenotype and the pharmacokinetics and pharmacodynamics of propranolol. Br J Clin Pharmacol 17:679–685. https://doi.org/10.1111/j.1365-2125.1984.tb02403.x
Walle T, Walle UK, Wilson MJ, Fagan TC, Gaffney TE (1984) Stereoselective ring oxidation of propranolol in man. Br J Clin Pharmacol 18:741–748. https://doi.org/10.1111/j.1365-2125.1984.tb02537.x
Yu L, Qian M, Liu Y, Yao T, Zeng S (2010) Stereoselective metabolism of propranolol glucuronidation by human UDP-glucuronosyltransferases 2B7 and 1A9. Chirality 22:456–461. https://doi.org/10.1002/chir.20765
Venkatakrishnan K, Obach RS (2005) In vitro-in vivo extrapolation of CYP2D6 inactivation by paroxetine: prediction of nonstationary pharmacokinetics and drug interaction magnitude. Drug Metab Dispos 33:845–852. https://doi.org/10.1124/dmd.105.004077
Kim J (2000) Pharmacokinetics and pharmacodynamics of the selective serotonin reuptake inhibitors, fluoxetine and paroxetine, during pregnancy and the nursing period. PhD Thesis, University of British Columbia, Vancouver
Acknowledgements
The authors thank Dr. William J. Jusko (State University of New York, Buffalo) for providing theophylline clinical PK data used for model verification, Dr Ruth Clayton for helpful comments and proofreading and Jessica Waite for prompt assistance in preparing the manuscript for submission.
Funding
No funding was received for the preparation of this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All authors are full-time employees of Certara UK Limited, Simcyp Division. The activities of Certara are supported by a consortium of pharmaceutical companies. The Simcyp Simulator is freely available, following completion of the training workshop, to approved members of academic institutions and other not-for-profit organizations for research and teaching purposes.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abduljalil, K., Pansari, A. & Jamei, M. Prediction of maternal pharmacokinetics using physiologically based pharmacokinetic models: assessing the impact of the longitudinal changes in the activity of CYP1A2, CYP2D6 and CYP3A4 enzymes during pregnancy. J Pharmacokinet Pharmacodyn 47, 361–383 (2020). https://doi.org/10.1007/s10928-020-09711-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-020-09711-2