
Abstract
The International Council for Harmonisation revised the E14 guideline through the questions and answers process to allow concentration-QTc (C-QTc) modeling to be used as the primary analysis for assessing the QTc interval prolongation risk of new drugs. A well-designed and conducted QTc assessment based on C-QTc modeling in early phase 1 studies can be an alternative approach to a thorough QT study for some drugs to reliably exclude clinically relevant QTc effects. This white paper provides recommendations on how to plan and conduct a definitive QTc assessment of a drug using C-QTc modeling in early phase clinical pharmacology and thorough QT studies. Topics included are: important study design features in a phase 1 study; modeling objectives and approach; exploratory plots; the pre-specified linear mixed effects model; general principles for model development and evaluation; and expectations for modeling analysis plans and reports. The recommendations are based on current best modeling practices, scientific literature and personal experiences of the authors. These recommendations are expected to evolve as their implementation during drug development provides additional data and with advances in analytical methodology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The International Council for Harmonisation (ICH) E14 Questions and Answers document was revised in December 2015 to allow for concentration-QTc (C-QTc) modeling to be used as the primary analysis for assessing the QTc interval (hereafter referred to as QTc) prolongation risk of new drugs [1]. There are several important implications of this revision on the design and analysis of thorough QT/QTc (TQT) studies. Because the C-QTc modeling approach utilizes data from all doses and time points, a reliable assessment of QTc prolongation can be based on smaller-than-usual TQT trials or be obtained during trials in early development not specifically targeted at QT effects. Thus, sponsors of pharmaceutical products now can either perform smaller TQT studies or use C-QTc modeling with high quality electrocardiogram (ECG) measurements in single- and/or multiple- dose escalation (SAD/MAD) studies during early-phase clinical development as an alternative to the central tendency by time point approach to meet the regulatory requirements of the ICH E14 guideline [2]. When utilizing a modeling approach for regulatory purposes, it is particularly important that the quantitative methods used be clearly described to enable reproducibility. It is expected that data sources and modeling details, including the structural model, assumptions, criteria for assessment of model robustness and goodness-of-fit, be adequately described in a pre-specified modeling analysis plan (MAP), and reported in a standardized format [3].
The E14 implementation working group did not provide the technical details on how to perform and report C-QTc modeling to support regulatory submissions. The rationale for this approach is that specific analysis methodologies are expected to evolve over time as pharmaceutical and regulatory scientists implement this approach across drugs with diverse pharmacokinetic (PK) and pharmacodynamic (PD) attributes. The objective of this White Paper is to propose recommendations for designing studies to use C-QTc modeling as the primary analysis, conducting a C-QTc analysis, and reporting the results of the analysis to support regulatory submissions.
Study design considerations for C-QTc analysis
The TQT study is intended to determine whether the drug has a threshold pharmacologic effect on cardiac repolarization, as detected by QTc prolongation. The threshold level of regulatory concern is around 5 ms as evidenced by an upper bound of the 95% confidence interval around the mean effect on QTc of 10 ms [2]. TQT study results are used to determine whether the effect of a drug on the QT/QTc interval in target patient populations should be studied intensively during later stages of drug development. In some cases, a well-designed and conducted QTc assessment based on C-QTc analysis in early phase 1 studies can be an alternative approach to a TQT study to reliably exclude clinically relevant QTc effects. The appropriateness of these data will not be generally known until later in the development program when the therapeutic dose level has been identified and the intrinsic and extrinsic factors that increase exposures to drug and active metabolites are known. In this section, we discuss general study design features that should be considered when assessing whether the data can be used in a C-QTc analysis to substitute for a TQT study. It is anticipated that this approach will not be applicable to all drugs, such as for drugs with substantial heart rate effects, active metabolites that inhibit cardiac ion channels or formulations that have a narrow range of concentrations. Descriptions of these challenging drugs are presented in Table 1.
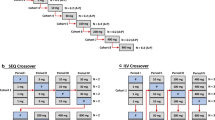
Study design
It is beyond the scope of this White Paper to provide all the study design features of early phase 1 clinical studies. SAD/MAD studies are generally acceptable for early QTc assessment. Commonly used designs are the sequential parallel group design (where the doses are gradually escalated, and after each dose administration and subsequent safety evaluation, a new cohort with new subjects is included and administered the next dose level) and the alternating panel crossover design (where dose escalation is alternated between 2 or more panels and each panel receives every other dose, and during the dosing of one cohort, the other cohort(s) are in washout). The choice of design is based on the study objectives, only one of which is the assessment of QTc prolongation. A placebo cohort should be used whenever possible to control for potential bias introduced by study procedures and to increase the power to exclude modest QTc effects in small-sized studies [4]. The general mechanisms to deal with potential bias and reduce study variability are discussed in the ICH E14 guidance for the TQT study and these are also applicable to the early phase 1 studies for QTc assessment (Supplemental Material, Table S1).
Baseline ECGs
Regardless of study design, time-matched baseline ECG recordings are generally not required in early phase 1 parallel studies because the inclusion of the placebo data allows for the detection of diurnal patterns in the QTc data. PK/PD simulations have shown that accounting for diurnal variability increases the power of the C-QTc analysis to exclude small mean QTc effects [4]. The collection of baseline ECG data over a wide range of heart rates on the day before dosing is recommended for drugs that meaningfully affect heart rate [5]. Although there is no consensus on the best approach to characterize the QT/QTc interval for these drugs, it is common to compute a subject-specific heart-rate-corrected QT interval derived from QT/RR pairs collected from baseline ECG recordings [6].
Sample size
Early phase 1 studies will not be specifically powered for the QTc assessment and will generally be under-powered for using the intersection union test by dose cohort [7, 8]. In general, typical SAD/MAD studies contain at least four dose cohorts, with each cohort having 4–8 subjects on drug and 2–4 subjects on placebo; and these are likely to be sufficient for early QTc assessment based on C-QTc analysis if the study is well conducted per Supplemental Material, Table S1 [9, 10]. Stochastic PK/PD simulations have shown that the false negative rate (i.e., falsely concluding no QTc prolongation) of C-QTc analyses is controlled at around 5% when the true effect is 10 ms in small-sized studies of 6–12 subjects with multiple measurements per subject [4, 11,12,13]. When simulations were performed using the same sample sizes and assuming no underlying QTc effect (placebo), the fraction of studies in which an effect above 10 ms could be excluded was above 85% [12, 13]. Power to exclude a 10-ms effect will be less for a drug with small true mean QTc effects (i.e., 3–5 ms) and will be influenced by variability in the QTc measurements.
Exposure margin
To ensure adequate QTc assessment, the exposure in early phase studies should be well above the maximum therapeutic exposure to cover the potential impact of intrinsic and extrinsic factors, including unanticipated factors, on drug exposure. There are several circumstances where C-QTc analysis of data from repeat doses of drug is recommended: for drugs that have significant PK accumulation of the parent or potentially clinically significant metabolite(s) in plasma on repeat dosing to steady state; if the exposure to the parent drug or metabolites(s) at the maximum tolerated single dose does not match or exceed the supratherapeutic exposure at steady state.
Positive control
A limitation of performing QTc assessment in early phase 1 studies is the lack of a positive control to demonstrate ECG assay sensitivity [14]. Without a separate positive control and thus a direct measure of assay sensitivity, the QTc response should be characterized at a sufficiently high multiple of the clinically relevant exposure [1]. The FDA’s Interdisciplinary Review Team is recommending that exposures are at least twice the highest clinically relevant exposure (i.e., the highest dose should give a mean Cmax that is twice the Cmax,ss obtained during metabolic inhibition with a concomitant drug or with renal or hepatic dysfunction) to obviate the positive control. If this exposure margin cannot be achieved in the early phase studies, a dedicated TQT study that includes a positive control may still be needed until a non-pharmacological approach for assay sensitivity has been validated [15,16,17].
Pooling data
Pooling data from multiple studies is not recommended in cases where differences in the study conditions may cause bias in results: (1) the study control procedures (e.g., placebo, food control) are different; (2) ECG acquisition and ECG measurement at baseline and during the treatment are different; or (3) study subjects are taking concomitant medications or with comorbid conditions that increase the variability in the QTc interval in one study but not the other. If there is a need to pool data from multiple studies to cover a wide range of doses/exposures or to increase the number of subjects exposed to drug at higher doses, it is important that similar clinical conduct and subject handling be performed in each study and that the ECG acquisition and ECG measurement approaches be similar (Supplemental Material, Table S1), as well as the bioanalytical assay. It is preferred that the concentration–response data come from related study protocols (e.g., SAD/MAD) to ensure that study procedures and timing of ECG measurements are similar across dose levels and placebo arms.
Table 2 summarizes the study design features that influence the decision to use C-QTc analysis of data collected in a phase 1 study to replace a TQT study. It is important that ECG quality, dose/exposure margin, sample size, timing of PK sample and ECG recordings and effects on HR are considered when planning to use C-QTc analysis in a phase 1 study (or pooled studies) that is intended to substitute for a TQT study.
Modeling approach
Objectives
Concentration-QTc analysis can serve as an alternative to the by-timepoint analysis or intersection–union test as the primary basis for decisions to classify the risk of a drug [1]. When C-QTc analysis is utilized as the primary basis for decisions to classify the risk of a drug, the upper bound of the two-sided 90% confidence interval for model-derived ΔΔQTc should be < 10 ms at the highest clinically relevant exposure (see [2, E14 Sect. 2.2.2]) to conclude that an expanded ECG safety evaluation during later stages of drug development is not needed [1]. A pre-specified linear mixed effects model is recommended as the primary analysis to exclude a 10-ms QTc prolongation effect. For drugs that prolong the QTc interval, however, the objectives of the C-QTc analysis are more exploratory and the pre-specified model may not be appropriate in this setting.
Baseline-adjusted QTc interval
For the pre-specified model, the preferred dependent variable in the model is the baseline-adjusted and Fridericia heart-rate-corrected QT interval (ΔQTcF) [13]. Unless drug-free QT data are collected in all subjects over a range of heart rates like the range of heart rates observed during treatment, the use of subject- and study-specific corrections is not generally recommended. The choice of which baseline correction method (predose versus time-matched) to use will depend on study design and should be pre-specified in the MAP. Predose baseline adjustment uses the average of QTc measurements obtained immediately prior to dosing as the baseline of each treatment period. Time-matched baseline adjustments use the corresponding predose QTc measurements collected prior to drug administration on Day-1. A time-matched baseline adjustment may be used when placebo data are not available to minimize the effect of diurnal variation in QTc [18].
Although the focus of this white paper is on the QTc interval, the same methodology can be applied to other ECG parameters such as heart rate (HR), PR interval, QRS complex, JTc interval, J − Tpeakc and Tpeak − Tend.
Pre-specified linear mixed effects model
The pre-specified linear mixed effect (LME) C-QTc model is used as the primary analysis. This model, while overparameterized (i.e., a model that contains parameters that may not influence the prediction), appropriately addresses the overall modeling objective. The default dependent variable is ΔQTcF; hereafter the dependent variable is referred to more generically as ΔQTc. The fixed effect parameters are intercept, slope, influence of baseline on intercept, treatment (active = 1 or placebo = 0) and nominal time from first dose (Table 3). Subject is included as an additive random effect on both intercept and slope terms.
In Eq. 1, \(\Delta QTc_{ijk}\) is the change from baseline in QTc for subject i in treatment j at time k; θ0 is the population mean intercept in the absence of a treatment effect; η0,i is the random effect associated with the intercept term θ0; θ1 is the fixed effect associated with treatment TRTj (0 = placebo, 1 = active drug); θ2 is the population mean slope of the assumed linear association between concentration and ΔQTc ijk ; η2,i is the random effect associated with the slope θ2; Cijk is the concentration for subject i in treatment j and time k; θ3 is the fixed effect associated with time; and θ4 is the fixed effect associated with baseline \(QTc_{i,j = 0}\); \(\overline{QTc}_{0}\) is overall mean of \(QTc_{ij0}\), i.e., the mean of all the baseline (= time 0) QTc values. It is assumed the random effects are normally distributed with mean [0,0] and an unstructured covariance matrix G, whereas the residuals are normally distributed with mean 0 and variance R.
In the above model, an unstructured covariance matrix is the preferred random effects covariance matrix because it does not impose constraints on the variances. Other random effect models based on the specific study design are also acceptable (e.g., with crossover designs [19]); however, over-parameterized random effects models may not converge and may need to be simplified for convergence to occur. Although this model is technically an error-in-variables problem, measurement error in C does not affect the estimation of the concentration slope unless the measurement error exceeds 30% [20].
Model parameters are to be estimated using maximum likelihood or restricted maximum likelihood approaches. There is no recommendation for preferred software. The MAP should specify approaches to handle non-convergence of the model, including any transformations of concentration data. To avoid unnecessary model-building steps, removing non-significant parameters from the model is generally not recommended [21]. It is not expected that the slope estimate will be materially influenced by an overparameterized model if there are sufficient data in the placebo group (see Supplemental Material, Effect of Model Overparameterization in QT Analyses).
The pre-specified model is recommended because it can be used for most study designs in healthy subjects (e.g., SAD, MAD and TQT studies). There are, however, situations when changes to the pre-specified model are needed to accommodate different data structures, such as when ΔΔQTc is used as the dependent variable in the model, placebo data are not available, or when the data are pooled from multiple studies. Considerations for changes to the pre-specified model under these scenarios are summarized in Table 4.
Model independent checks of assumptions using exploratory plots
Basic assumptions made in the modeling process can efficiently be evaluated using simple graphics as summarized below with details provided in Table 5. If the following modeling assumptions are not supported by the data, the model development for adapted C-QTc model is recommended.
-
Assumption 1: No drug effect on HR One consideration is the potential of a drug to significantly increase or decrease HR. Although there is no consensus on the specific threshold effect on HR that could influence QT/QTc assessment, mean increases or decrease > 10 bpm have been considered problematic [5]. The time course of mean ΔHR/ΔΔHR effects by treatment is a useful evaluation (Fig. 1).
Fig. 1 
Evaluation of drug induced effect on heart rate by dose: a time course of the mean change from baseline in heart rate and b the mean change from baseline placebo-adjusted heart rate
-
Assumption 2: QTc interval is independent of HR QTcF is usually a sufficient correction method for drugs with insignificant effects on HR and evaluation of this correction method is not needed. If an individualized heart rate correction method is used, the appropriateness of the selected method should be assessed to determine if QTc is independent of HR using drug-free data (Fig. 2).
Fig. 2 
Evaluation of the heart rate corrected QT interval: a scatterplot of QTcF and RR intervals by treatment and b QTcF-RR quantile plot (with quintiles) with linear mixed effects line and 95% confidence interval
-
Assumption 3: No time delay between drug concentrations and ΔQTc Concordance, or lack thereof, in the time course of drug concentrations and ΔΔQTc − ΔQTc corrected for spontaneous diurnal variation as seen in a placebo group—can be evaluated by examining the mean concentration and QTc profiles by dose level and time (Fig. 3a–f). If the time course of ΔΔQTc is concordant with the PK profile, the default model assumption of a direct temporal relationship between drug concentration and QTc effect can be supported (Fig. 3, left panels). If, however, there is a delay between peak concentration and peak QTc or QTc effect (Fig. 3, right panels), the analyst must consider the possibility of a delayed temporal effect and the model applied should account for this delay (see “Model development for adapted C-QTc models”). An isolated outlying mean value of ΔΔQTcF, e.g., in the late elimination phase of the drug, need not be an indication for a systematic delay between concentration and effect.
Fig. 3 
Evaluation of PK/PD hysteresis using exploratory plots with examples of a direct effect in left panels and a 1-h delayed-effect in right panels: a, b time course of mean and 90% CI ΔΔQTcF and c, d drug concentration; e, f mean ΔΔQTc and concentration connected in temporal order by dose; f, g scatter plot of paired ΔQTc and concentration data with loess smooth line and 95% confidence intervals (shading) and linear regression line (solid line) (Color figure online)
-
Assumption 4: Linear C-QTc relationship The adequacy of using a linear model can be assessed by the concentration-ΔQTc plot incorporating a trend line (e.g., loess smooth or linear regression, Fig. 3g, h). The trend line does not reflect a model fit of data, but rather is used to detect drug effect, and when a drug effect is detected, whether there are major violations to the linear assumption. If data are pooled from multiple studies, trend lines are to be displayed for each study and pooled across studies.



C-QTc quantile plots are an effective format for displaying relationships that may be difficult to identify in the presence of highly variable data and a low signal-to-noise ratio [22]. The C-QTc quantile plots used in the examples are generated by binning the independent variable (e.g., concentrations) into quantiles (bins with equal number of observations, typically deciles) and plotting the local median or midpoint of the observations in each of the independent variable bins against the corresponding local mean dependent variable (e.g., ΔQTc or ΔΔQTc and associated 90% confidence).
Model development for adapted C-QTc models
If the exploratory plots indicate the modeling assumptions are not met, additional modeling steps are recommended to determine objectively the appropriate C-QTc model. When apparent differences in the time course and/or distribution of HR between on- and off-drug conditions are observed in exploratory plots, other approaches to evaluate QT/QTc should to be considered as summarized in [5].
It is important that the drug effect model adequately describe the observed concentration and ΔQTc relationship to obtain reliable estimation of the degree of prolongation at doses/concentrations of interest. The drug effect models routinely tested are the linear and Emax families of models; however, other types of PD models can be explored to optimize the model fit. Simpler models are preferred over more complex models when statistically justified. Logarithmic transformation of the concentration data is not recommended because the proposed modeling approach uses placebo concentrations that are set to zero. Because the results of the analysis will be critical to assessment of risk, the model and methods for model evaluation and selection need to be pre-specified in a MAP to limit bias. Model selection should be based on the pre-specified objective criteria (e.g., objective function value, Akaike information criteria (AIC), level of statistical significance, goodness of fit plots, standard error in model parameters) and follow standard modeling practices including sensitivity analyses and model qualification [3, 23].
If exploratory plots indicate a delay between drug concentration and the effect on QTc (Fig. 3, right panels), the presence of active metabolites should be considered and models including the concentration of these metabolites explored. For example, QT, QRS, and PR interval prolongations with Anzemet appear to be associated with higher concentrations of its active metabolite, hydrodolasetron [24]. If metabolites do not explain the delay between QTc-effect and the concentration of the parent drug, a PK/PD model with a separate effect compartment may, at times, be helpful [25]. There are some investigations in the use of metrics or tests for the absence of hysteresis between PK and QTc, but there is little experience on their applicability in practice [26, 27].
In most cases, covariate analysis (e.g., effect of sex, weight) will not be routinely performed with C-QTc models based on data from healthy volunteers. There may be, however, instances where covariate identification is of interest. The method for covariate selection should be pre-specified in the MAP, including methods for handling highly correlated covariates, such as baseline QTc and sex.
Model evaluation
Goodness-of-fit (GOF) plots, as described in Table 5, should be presented for the final C-QTc model, and when relevant for key stages during model development (see Supplemental Material, Case Study of a Misspecified Model). It is recommended to choose GOF plots appropriate for the analysis, as the value of different GOF plots may be dependent on the situation. Both scatterplots and quantile plots are useful representations of the residuals for continuous model parameters (e.g., concentrations, baseline QTc) and boxplots are useful for categorical parameters (e.g., time, treatment). A C-QTc quantile plot of observed data overlaid with the model predictions (Fig. 4a, b) is another visual assessment of how well the model fits the data.

Evaluation of the final model for correctly specified model (left panels) and misspecifed model (right panels): a, b ΔQTc versus drug concentration quantile plot with model slope and 90% CI; and c, d ΔΔQTc versus concentration with associated predictions shown by arrows at mean Cmax by dose. In the C-QTc quantile plot, it should be noted that with a model including time as factor, the regression line y = Θx + t with slope Θ and treatment effect t will not fit the observed data if the average time effect is not zero. To obtain a visual fit, either the regression line or the observed data (ΔQTc) need to be adjusted for the time effect (Color figure online)
Model parameters are to be presented in tabular format showing the estimate, standard error of the estimate, p value and 95% confidence interval. Model parameters that are not supported by the observed data will be poorly estimated and their 95% confidence intervals will include zero. Because an over-parameterized model may at times have lower AIC values compared to a simpler model, it is important that the MAP describes specific criteria for choosing the appropriate model. Parameter estimates should also be evaluated for evidence of model misspecification (Supplemental Material, Table S2). Similarly, a large significant treatment effect may indicate a misspecified model as illustrated in the Supplemental Material, Case Study of a Misspecified Model, and shown by PK/PD simulations [13].
Estimation of model-derived ΔΔQTc at concentration(s) of interest
The final C-QTc model should be used to compute the ΔΔQTc at concentrations of interest, such as concentrations representing a therapeutic dose in a patient population and the expected increased concentrations associated with drug interactions, impaired hepatic or renal function, and in patients with polymorphisms in CYP enzymes. Because the C-QTc models are data-driven and empirical PD models are used to describe the observed data, it is strongly recommended that the model not be extrapolated to concentrations that fall outside the range of observed concentrations used to generate the model. For decision making, the concentration of interest will be derived from the drug development program and, therefore, can be treated as a prediction variable without concerns of uncertainty.
For the pre-specified C-QTc model, the change from baseline QTc adjusted for placebo (ΔΔQTc) is the difference between the model-derived ΔQTc at concentration of interest and model-derived ΔQTc for placebo (concentration = 0).
where ∆QTc(C,trt=active) is the ΔQTc at concentration from the final C-QTc model using ΔQTc as the dependent variable, C is the concentration of interest.
In the simplest case and when using the pre-specified linear model, the mean and two-sided 90% CI for ΔΔQTc at concentrations of interest (i.e., Cmax at specific dose level) are computed from Eqs. 3–5, where “Est” refers to an estimate of the true parameter from the model fit.
where \(\theta_{1,Est}\) is the estimated treatment-specific intercept; \(\theta_{2,Est}\) is the estimated slope, \(var(\theta_{1,Est} )\) is the variance of the treatment-specific intercept; \(var(\theta_{2,Est} )\) is the variance of the slope; \(cov(\theta_{1,Est} \theta_{2,Est} )\) is the covariance of the intercept and slope; t is the critical value determined from the t-distribution; DF is the degrees of freedom; SE is the standard error; and CI is the confidence interval.
For adapted C-QTc models that have different structures (e.g., nonlinear models or linear models with interaction terms), the mean and 90% CI for ΔΔQTc can be computed by non-parametric bootstrap methods with subject identifier used as the unit for resampling. Resampling should be stratified by dose level within the active treatment and the placebo subjects. Model parameters and ΔΔQTc (Eq. 2) are determined from each of the replicate bootstrapped datasets. Two-sided 90% CIs are computed from the 5th and 95th percentile of the rank-ordered ΔΔQTc values from all replicates and bias correction may be applied.
Reporting of C-QTc modeling results
Modeling analysis plan
A detailed plan should be prospectively written for the C-QTc analysis. The plan should include the following:
-
The objective(s) of the analysis.
-
A brief description of the study (or studies) from which the data originate and, if pooling data, the rationale for pooling data and a discussion of the similarities with respect to clinical conduct, subject handling and ECG acquisition, ECG measurement approaches, and bioanalytical assay.
-
The key features of the data to be analyzed (e.g., how many studies, subjects, dose levels, time-matched PK/ECG samples).
-
A brief description of ECG acquisition and measurement, including the use of core ECG laboratory, analysis approach, and blinding procedures of ECG readers.
-
The procedures for data transformations, handling missing data, outlying data and concentration data below the limitation of quantification.
-
The general modeling aspects (e.g., software, estimation methods).
-
The graphical data exploration of model assumptions (as described in Table 5).
-
The dependent variable including methods for heart rate correction and baseline-correction.
-
The C-QTc models to be tested, including alternative models to be used in cases of non-convergence, PK/PD hysteresis, nonlinearity, et cetera.
-
If pooling data, the objective criteria for testing heterogeneity.
-
The covariate models to be tested together with a rationale for including these covariates.
-
The criteria to be used for selection of models during model building and inclusion of covariates (e.g., objective function value, AIC, level of statistical significance, GOF plots, standard error in model parameter, inter-individual variability, clinical relevance).
-
The methods to be used for model-based predictions and the rationale for choosing the exposure of interest.
There may be cases where the selection of a specific model can only be made once data are available. The principles governing this selection are clearly pre-specified. The MAP can be a stand-alone document or incorporated into the statistical analysis sections of the protocol.
Modeling results
Recommendations for information to be reported are summarized in Table 6 and are based on European Medicines Agency’s Guideline on reporting the results of population pharmacokinetic analyses [28] and recommendations by European Federation of Pharmaceutical Industries and Associations’ working group on Model-Informed Drug Discovery and Development [3]. The modeling output can be documented in a stand-alone report or within specific report sections in the cardiac safety report or clinical study report.
Summary
This White Paper provides recommendations on how to plan and conduct definitive QTc assessment of a drug using C-QTc modeling in early phase clinical pharmacology and TQT studies. The recommendations are based on current best modeling practices, examples in scientific literature, and personal experiences of the authors. These recommendations are expected to evolve as their implementation during drug development provides additional data and with advances in analytical methodology.
A critical recommendation is a pre-specified LME model as the primary analysis to exclude a 10-ms QTc prolongation effect. A pre-specified model minimizes bias, as subjectivity in model selection could favor a model that inflates the type I error. Pre-specification allows for standardization of the analysis across industry and regulatory regions and offers several additional benefits, including providing transparency in modeling expectations, increasing the reproducibility of results, and streamlining the review process. It is expected that the pre-specified LME model will be suitable for most the drugs evaluated, with potential exceptions described in Table 1. The intention of the recommended modeling approach is to simplify and increase the objectivity of traditional PK/PD modeling process by utilizing the pre-specified model and, if drug effect is detected, optimizing the drug-effect model using PK/PD modeling principles. In this scenario, it is extremely important that the model qualification and selection process follows the MAP to avoid bias. It is possible that the model-derived ΔΔQTc of the final model will be lower than one derived from the pre-specified model. To minimize inconsistent interpretation, the reporting of these results should focus on (1) the final model selection process and how well the final model describes the data; (2) the difference between models in the model-derived ΔΔQTc at exposure of interest; and (3) whether the exposure of interest is therapeutic or supratherapeutic.
To use the C-QTc modeling analysis of data from a phase 1 study as an alternative to a TQT study, a critical element is that the QT response be evaluated at a sufficiently high multiple of the clinically relevant exposure to waive the need for assessing assay sensitivity with an active control. A large exposure margin provides confidence that small QTc effects at clinically relevant exposures have not been missed in the evaluation. Other important design elements that are common to TQT studies should be included in the phase 1 design. Careful subject handling and robust ECG acquisition and blinded measurement are necessary to minimize bias and variability. Time-matched PK samples and ECG measurements should cover the inter-dosing interval and include time of peak concentrations of parent and metabolites and be collected for at least 24 h following a single dose. For a drug with a long half-life of parent or active metabolites, there needs to be ECG measurements taken when peak concentrations are achieved or at earlier times at higher doses that give the maximum concentrations achieved over time. The number of subjects on placebo and pooled doses should provide sufficient power to exclude a 10-ms QTc prolongation effect using C-QTc analysis.
Overall, the recommendations within the White Paper provide opportunities for increasing efficiencies in this safety evaluation. Given the decade of experience using C-QTc model in drug development and regulatory decisions, the recommended modeling approach is not anticipated to compromise detection of drugs with QT liability.
Change history
12 January 2018
The original version of this article unfortunately contained an error in Equation 1 under the section “Pre-specified linear mixed effects model”. The correct equation has given below.
Abbreviations
- AIC:
-
Akaike information criteria
- C:
-
Concentration
- CI:
-
Confidence intervals
- Cmax :
-
Maximum concentration
- C-QTc:
-
Concentration-QTc
- ΔHR:
-
Baseline-corrected heart rate
- ΔQTc:
-
Baseline-corrected QTc interval
- ΔΔQTc:
-
ΔQTc interval corrected for placebo
- ECG:
-
Electrocardiogram
- Emax :
-
Maximum effect
- ER:
-
Extended release
- GOF:
-
Goodness-of-fit
- hERG:
-
Human ether-a-go-go-related gene
- HR:
-
Heart rate
- ICH:
-
International Council for Harmonization
- IR:
-
Immediate-release
- ms:
-
Milliseconds
- LME:
-
Linear mixed effects
- MAD:
-
Multiple-ascending dose
- MAP:
-
Modeling analysis plan
- MD:
-
Multiple dose
- PK:
-
Pharmacokinetic
- PD:
-
Pharmacodynamics
- QT:
-
QT interval on ECG
- QTc:
-
QT interval corrected for heart rate
- QTcF:
-
Fridericia corrected QT interval
- SAD:
-
Single-ascending dose
- SD:
-
Single dose
- TQT:
-
Thorough QT/QTc
References
ICH E14 Guideline (2015) The clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. Questions & answers (R3). http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Q_As_R3__Step4.pdf
ICH E14 Guideline (2005) The Clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non-antiarrhythmic drugs. http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf
Marshall SF et al (2016) Good practices in model-informed drug discovery and development: practice, application, and documentation. CPT Pharm Syst Pharmacol 5(3):93–122
Ferber G, Zhou M, Darpo B (2015) Detection of QTc effects in small studies—implications for replacing the thorough QT study. Ann Noninvasive Electrocardiol 20(4):368–377
Garnett CE et al (2012) Methodologies to characterize the QT/corrected QT interval in the presence of drug-induced heart rate changes or other autonomic effects. Am Heart J 163(6):912–930
Malik M, Hnatkova K, Batchvarov V (2004) Differences between study-specific and subject-specific heart rate corrections of the QT interval in investigations of drug induced QTc prolongation. Pacing Clin Electrophysiol 27(6 Pt 1):791–800
Zhang J, Machado SG (2008) Statistical issues including design and sample size calculation in thorough QT/QTc studies. J Biopharm Stat 18(3):451–467
Malik M et al (2008) Near-thorough QT study as part of a first-in-man study. J Clin Pharmacol 48(10):1146–1157
Darpo B et al (2015) Results from the IQ-CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin Pharmacol Ther 97(4):326–335
Nelson CH et al (2015) A quantitative framework to evaluate proarrhythmic risk in a first-in-human study to support waiver of a thorough QT study. Clin Pharmacol Ther 98(6):630–638
Darpo B et al (2014) The IQ-CSRC prospective clinical Phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?”. Ann Noninvasive Electrocardiol 19(1):70–81
Ferber G, Lorch U, Taubel J (2015) The power of phase i studies to detect clinical relevant QTc prolongation: a resampling simulation study. Biomed Res Int 2015:293564
Garnett C et al (2016) Operational characteristics of linear concentration-QT models for assessing QTc interval in the thorough QT and I clinical studies. Clin Pharmacol Ther 100(2):170–178
Zhang J (2008) Testing for positive control activity in a thorough QTc study. J Biopharm Stat 18(3):517–528
Ferber G et al (2017) Can bias evaluation provide protection against false-negative results in QT studies without a positive control using exposure-response analysis? J Clin Pharmacol 57(1):85–95
Taubel J, Ferber G (2015) The reproducibility of QTc changes after meal intake. J Electrocardiol 48(2):274–275
Taubel J, Fernandes S, Ferber G (2017) Stability of the effect of a standardized meal on QTc. Ann Noninvasive Electrocardiol. https://doi.org/10.1111/anec.12371
Zhang J, Dang Q, Malik M (2013) Baseline correction in parallel thorough QT studies. Drug Saf 36(6):441–453
Mehrotra DV et al (2017) Enabling robust assessment of QTc prolongation in early phase clinical trials. Pharm Stat 16(3):218–227
Bonate PL (2013) Effect of assay measurement error on parameter estimation in concentration-QTc interval modeling. Pharm Stat 12(3):156–164
Gastonguay MR (2004) A full model estimation approach for covariate effects: inference based on clinical importance and estimation precision. AAPS J 6(S1):W4354
Tornoe CW et al (2011) Creation of a knowledge management system for QT analyses. J Clin Pharmacol 51(7):1035–1042
FDA (1999) Guidance for industry: population pharmacokinetics. U.S.D.O.H.H. Services. http://www.fda.gov/downloads/Drugs/…/Guidances/UCM072137.pdf
FDA (2010) Drug safety communication: abnormal heart rhythms associated with use of Anzemet (dolasetron mesylate). 2010 [cited 11 July 2016]. http://www.fda.gov/Drugs/DrugSafety/ucm237081.htm
Holford NH et al (1981) The effect of quinidine and its metabolites on the electrocardiogram and systolic time intervals: concentration–effect relationships. Br J Clin Pharmacol 11(2):187–195
Glomb P, Ring A (2012) Delayed effects in the exposure-response analysis of clinical QTc trials. J Biopharm Stat 22(2):387–400
Wang JX, Li WQ (2014) Test hysteresis in pharmacokinetic/pharmacodynamic relationship with mixed-effect models: an instrumental model approach. J Biopharm Stat 24(2):326–343
EMA (2017) Guideline on reporting the results of population pharmacokinetic analyses. 2007 [cited 2016]. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003067.pdf
Bonate PL (2013) The effects of active metabolites on parameter estimation in linear mixed effect models of concentration-QT analyses. J Pharmacokinet Pharmacodyn 40(1):101–115
Sinclair K et al (2016) Modelling PK/QT relationships from phase I dose-escalation trials for drug combinations and developing quantitative risk assessments of clinically relevant QT prolongations. Pharm Stat 15(3):264–276
Zhu H et al (2010) Considerations for clinical trial design and data analyses of thorough QT studies using drug–drug interaction. J Clin Pharmacol 50(10):1106–1111
Mehta R et al (2016) Concentration-QT analysis of the randomized, placebo- and moxifloxacin-controlled thorough QT study of umeclidinium monotherapy and umeclidinium/vilanterol combination in healthy subjects. J Pharmacokinet Pharmacodyn 43(2):153–164
Sarapa N, Britto MR (2008) Challenges of characterizing proarrhythmic risk due to QTc prolongation induced by nonadjuvant anticancer agents. Expert Opin Drug Saf 7(3):305–318
Isbister GK, Friberg LE, Duffull SB (2006) Application of pharmacokinetic-pharmacodynamic modelling in management of QT abnormalities after citalopram overdose. Intensive Care Med 32(7):1060–1065
Johannesen L et al (2014) Differentiating drug-induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin Pharmacol Ther 96(5):549–558
Johannesen L et al (2016) Late sodium current block for drug-induced long QT syndrome: results from a prospective clinical trial. Clin Pharmacol Ther 99(2):214–223
Taubel J et al (2012) Shortening of the QT interval after food can be used to demonstrate assay sensitivity in thorough QT studies. J Clin Pharmacol 52(10):1558–1565
Taubel J et al (2013) Insulin at normal physiological levels does not prolong QT(c) interval in thorough QT studies performed in healthy volunteers. Br J Clin Pharmacol 75(2):392–403
Shah RR et al (2015) Establishing assay sensitivity in QT studies: experience with the use of moxifloxacin in an early phase clinical pharmacology study and comparison with its effect in a thorough QT study. Eur J Clin Pharmacol 71(12):1451–1459
Huh Y, Hutmacher MM (2015) Evaluating the use of linear mixed-effect models for inference of the concentration-QTc slope estimate as a surrogate for a biological QTc model. CPT Pharm Syst Pharmacol 4(1):e00014
Disclaimer
The views presented in this article are the personal opinions of the authors and do not reflect the official views of their respective organizations.
Author information
Authors and Affiliations
Corresponding author
Additional information
The original version of this article was revised: The error in Equation 1 has been corrected.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Garnett, C., Bonate, P.L., Dang, Q. et al. Scientific white paper on concentration-QTc modeling. J Pharmacokinet Pharmacodyn 45, 383–397 (2018). https://doi.org/10.1007/s10928-017-9558-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10928-017-9558-5