
Abstract
The mouse mammary gland is a complex tissue that proliferates and differentiates under the control of systemic hormones during puberty, pregnancy and lactation. Once a highly branched milk duct system has been established, during mid/late pregnancy, alveoli, little saccular outpouchings, sprout all over the ductal system and differentiate to become the sites of milk secretion. Here, we review the emerging network of the signaling pathways that connects hormonal stimuli with locally produced signaling molecules and the components of intracellular pathways that regulate alveologenesis and lactation. The powerful tools of mouse genetics have been instrumental in uncovering many of the signaling components involved in controlling alveolar and lactogenic differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
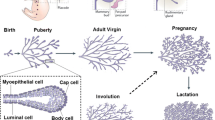
The mammary gland is the only organ that undergoes most of its development postnatally. During embryogenesis, a rudimentary ductal system develops. Under the influence of systemic hormones at puberty the ducts begin to expand into the surrounding stroma, the mammary fat pad, (Fig. 1a). With repeated estrous cycles and during pregnancy, the complexity of the ductal system increases through the addition of side branches (Fig. 1b; [1]). Finally, at mid/late pregnancy (day 14.5 in 129SV and C57Bl6 background), alveoli, form all over the ductal system (Fig. 1c). Alveoli are anatomical structures that have the form of a hollow cavity. They are also found in the terminal portions of other secretory organs such as the salivary gland and the pancreas and form grape-like structures at the end of bronchioles in the lung. The mammary alveoli develop into clusters by day 16.5 of pregnancy, and are dilated by the pressure of the secretions produced by the epithelial cells at the end of pregnancy (Fig. 1d; [1]).

Mouse mammary gland development during puberty, pregnancy and lactation, shown are whole mount micrographs. At the onset of puberty the rudimental ductal system invades and eventually fills the fat pad (a). With repeated estrous cycles and during the first half of pregnancy side branches form (b). During mid pregnancy the alveolar structures sprout from the expanded ductal tree (c). During lactation, alveoli are fully matured with distended ductal lumen and secrete milk (d). Hematoxylin and eosin stained section of mammary gland at day 14.5 of pregnancy. Note the presence of intracellular lipid droplet characteristic of this stage (e). The section of lactating mammary gland, arrow indicates the alveolar lumen filled with lipid droplets (f).
This last stage of mammary gland morphogenesis, alveologenesis, is closely intertwined with the functional differentiation of the mammary epithelium called lactogenesis. Two stages of lactogenesis are distinguished. Stage 1 of lactogenesis [2] begins at mid-pregnancy and involves increased and sustained expression of genes involved in the synthesis of different milk proteins such as β-casein, lactalbumin and WAP. Expression of various milk protein genes increases transiently during the estrous cycles [3]. Morphologically, stage 1 is characterized by intracellular lipid droplets (Fig. 1e). The second stage of lactogenesis occurs around parturition; expression of milk protein genes increases further, tight junctions between alveolar cells close, and cytoplasmic lipid droplets and casein move to the alveolar lumen ([4, 5]; Fig. 1f).
The observation that an early full-term pregnancy protects against breast cancer [6] has stimulated efforts to understand the events leading to full differentiation of the mammary gland. Many of the pathways involved in alveologenesis and lactogenic differentiation have been uncovered in recent years using genetically engineered mouse strains [7, 8], whereas the mechanisms underlying alveologenesis in the pancreas and salivary gland are largely unexplored.
Initially, gene targeting was used to delete genes in the mouse germ line [9]. The analysis of the role of a given gene product in the mammary gland can be confounded by systemic effects of gene inactivation. To circumvent this problem, transplantation techniques are used; mammary epithelium and/or mammary gland stroma, the fat pad, from donors of different genotypes are surgically recombined within wild-type (wt) host [10, 11]. Another caveat of the classical gene targeting approach is that the role of a gene at a specific stage cannot be discerned if the deletion has deranged earlier stages of development. This problem has been elegantly solved with the generation of inducible/conditional mouse mutant models. For example, during alveologenesis and lactogenic differentiation, genes can be specifically deleted by flanking the alleles with loxP binding sites. These sites are recognized by the Cre recombinase which can be expressed under the whey acidic protein (WAP) promoter [12] that is expressed in mammary epithelial cells undergoing secretory differentiation during pregnancy. The use of WAP and other promoters, for example MMTV, to drive the specific expression of the Cre recombinase has been instrumental in defining the role of numerous gene products in the mammary epithelium. Furthermore, such work has revealed that the organism is extremely plastic and able to compensate for germ-line mutations throughout development. Thus, in the case of X-linked inhibitor of apoptosis protein (XIAP) [13] as well as the transcription factor LMO4 [14] (see below), germ line deletion of these genes did not affect mammary gland development. In contrast, when either of these genes was deleted specifically during pregnancy milk secretion was impaired [13].
Hormonal Control of Alveologenesis and Lactogenic Differentiation
A series of systemic factors influence the mammary gland. Among them are the ovarian hormones estrogen and progesterone, pituitary prolactin, placental lactogens as well as the metabolic hormones, thyroid hormone, growth hormone (GH) and corticosteroids. In recent years evidence has accumulated that the mammary gland itself can secrete hormones such as prolactin [15], GH [16], leptin [17] and parathyroid hormone related peptide (PTHrP) [18]. PTHrP reaches the bloodstream and is involved in triggering calcium release from the bone during lactation. The local secretion of prolactin, was shown to influence mammary epithelial cell proliferation by an auto/paracrine mechanism [19], and it is generally held that GH and leptin also act within the mammary gland.
Adrenal glucocorticoids exert their biological activity by binding to their cognate receptors, glucocorticoid receptor (GR) and mineralocorticoid receptor. Cell culture experiments have long implicated corticosteroids as lactogenic factors [20]. Surprisingly, a recently developed mouse model in which the epithelial GR was deleted during pregnancy revealed that the receptor is dispensable for milk production but required for proliferation of mammary epithelial cells in vivo [21]. When the GR gene was deleted in the mammary epithelium by means of a WAP–Cre transgene, the cells showed reduced proliferation at day 14.5 of pregnancy but increased proliferation after parturition [21]. The mechanisms by which corticosteroids affect epithelial proliferation are unclear.
Another metabolic hormone, thyroid hormone, has also been shown to be important for lactogenesis [22], but whether it exerts its effect directly on the mammary gland or indirectly by regulating other signaling pathways is currently unclear.
Estrogen acts via two receptors, estrogen receptors α and β, (ERα and ERβ) [23, 24], both of which are expressed in the mammary gland [25–27]. ERα deficient mice are completely infertile [28]. Tissue recombination experiments revealed that ERα signaling is required in the mammary epithelium for ductal elongation during puberty [29]. When ERα deficient epithelium was grafted to cleared fat pads of wildtype hosts, expression of milk protein genes were induced during pregnancy [29], suggesting that ERα is not required for transcriptional response to lactogenic stimuli. ERβ deficient virgin females have ovarian defects, the rare mutants that got pregnant and lactated successfully had more dilated alveoli compared to wildtype animals [30]. Tight junctions between luminal epithelial cells were impaired, and extracellular matrix and basal lamina were reduced [30]. Whether this phenotype reflects a role of ERβ signaling during mammary epithelial differentiation, or whether it is a consequence of endocrine disturbances in the ERβ mutant mice needs further clarification.
Progesterone actions are mediated by two protein isoforms of the progesterone receptor PRA and PRB that are derived by transcription from two distinct promoters of the same gene. The longer B-form has an amino terminal extension containing a transactivation function that accounts for differential coactivator recruitment between the two forms. Tissue recombination studies with progesterone receptor (PR) knockout tissues [31] revealed that epithelial PR signaling is required for side branching and alveologenesis [10]. Elegant gene targeting experiments in which either the PRA or the PRB isoform were inactivated in the germ line revealed that deletion of the longer PRB form phenocopied the complete PR mutant mammary gland, whereas absence of the shorter PRA form did not impair mammary gland development [32], indicating that PRB is the predominant mediator of progesterone action in the mammary gland.
Other studies have demonstrated that PR is required only in a subset of mammary epithelial cells during development. Using mammary transplantation techniques, these studies further demonstrated that WT epithelium, placed adjacent to PR−/− epithelium, can mediate alveologenesis and lactogenic differentiation in the knockout tissue, arguing for a paracrine signaling function for PR in the gland during alveologenesis [10].
The two ovarian hormones, estrogen and progesterone orchestrate the unfolding of the ductal tree thereby setting the stage for alveolgenesis. The metabolic hormones appear to reinforce different stages of mammary gland development. The master controller of alveologenesis and lactogenic differentiation, however, is the pituitary polypeptide hormone, prolactin. In the absence of epithelial prolactin signaling, a fully branched ductal system develops but alveologenesis fails to occur and the mammary epithelium does not undergo secretory differentiation [33]. Indeed, females heterozygous for the inactivated PrlR allele, show a retardation in alveolar development and fail to feed their first litter [34]. The central importance of this signaling cascade is further underlined by the finding that loss of principal downstream mediators, JAK2 and STAT5a/b [35–38] and negative modulators, such as SOCS1 and 2, all have effects on alveologenesis (see below).
Not only is the Prolactin/JAK2/STAT5 cascade crucial for the functional differentiation of the mammary gland, but it also controls related events in other organs. Acting on pancreatic β-cells, prolactin triggers cell proliferation and insulin secretion thereby ensuring a series of metabolic adjustments required for milk secretion [39]. Hypothalamic neuroendocrine dopaminergic neurons increase dopamine turnover in response to prolactin. They release dopamine, which subsequently inhibits prolactin release from the anterior pituitary, functioning as a negative feedback loop [40].
Auto- and Paracrine Factors
Several local factors have been implicated in alveologenesis. Insulin-like growth factor 2 (IGF-2) was identified in an expression screen for prolactin-dependent genes in vivo and shown to be induced in primary mouse mammary epithelial cells by prolactin [41]. Analysis of IGF-2 deficient mice and ectopic expression of IGF-2 in PrlR−/− epithelia suggested that IGF-2 is involved specifically in alveolar morphogenesis, but not in the secretory differentiation of mammary epithelial cells (see Fig. 2).

Model of the signaling network that controls alveologenesis and lactogenic differentiation in the mouse mammary gland. The model is based on studies of different mouse mutant models discussed in the text.
Receptor activator of NFκB ligand, (RANKL) is a cytokine long known to be essential for the maturation of antigen-presenting cells and osteoclasts. Deletion of RANKL or its receptor RANK from the mammary epithelium blocked alveologenesis and lactogenic differentiation [42]. The RANKL promoter region contains a γ-interferon activation sequence (GAS) that conferred a STAT5 and JAK2 dependent prolactin response in reporter assays consistent with RANKL being a prolactin target gene [43]. In vivo, both progesterone [32, 41] and prolactin [43] have been linked to RANKL transcriptional control. The importance of different downstream signaling events is currently unclear. In the mammary epithelia of RANKL−/− mice, activation of STAT5 and ERK1/ERK2 was unaffected but AKT phosphorylation was decreased [42]. Based on these observations, together with their findings that apoptosis was increased and proliferation decreased in mammary epithelia of pregnant RANKL−/− mice, Fata et al. proposed that RANKL acts by inhibiting apoptosis via activation of the PI3kinase pathway. Cao et al. showed that IKKα is an important downstream mediator of RANKL and that RANKL affects proliferation via NFκB/Cyclin D1 [44]. However, biochemical evidence demonstrating the induction of Cyclin D1 by RANKL in the mammary epithelium is still missing [41, 45, 46]. And, recently the observation was made that mammary epithelial cells of RANKL−/− mice have normal cyclin D1 expression [45, 46]. Thus, the mechanism by which RANKL/RANK signaling contributes to alveologenesis and lactogenic differentiation warrants further studies.
Another locally produced factor important to both alveologenesis and lactogenic differentiation is the epidermal growth factor (EGF) family member, heregulin; more specifically, the HRG1 isoform expressed in the mammary gland, HRGα. Deletion of HRGα selectively impaired alveologenesis and lactogenic differentiation. The number of alveoli in HRGα deficient females was decreased, they were less distended and milk gene expression was reduced. Nevertheless, mutant mothers, were able to feed their pups [47], possibly because other ErbB ligands compensate for the absence of HRGα. Indeed, deletion of a conditional allele of the receptor, ErbB4, during pregnancy with a WAP–Cre transgene impairs alveologenesis and lactogenic differentiation more severely with only 20% of the offspring surviving [47]. The other cognate HRGα receptor, ErbB2, was inactivated by ectopic expression of a dominant negative mutant and resulted in a similar phenotype [48] consistent with the HRG/ErbB pathway being important in both alveologenesis and lactogenic differentiation.
Intracellular Signal Transduction
How does the cell react to all the stimuli it receives and how does it integrate the different inputs? The intracellular events are best understood in the case of prolactin signaling. Binding of prolactin triggers receptor dimerization. As a result, janus-2 kinase (JAK2) is recruited and activated. The activated JAK2, in turn, phosphorylates both the prolactin receptor and signal transducer and activator of transcription 5 (STAT5). Two STAT5 proteins exist, STAT5a and STAT5b, that share over 90% identity and diverge only in their carboxyl terminus. STAT5a is the predominant form in the mammary gland. Once activated, STAT5 dimerizes and translocates to the nucleus where it activates the transcription of target genes, such as different milk protein genes that contain GAS (γ-interferon activation sequence) in their promoters [49]. The suppressors of cytokine signaling (SOCS) are also induced by STATs and they negatively regulate cytokine signaling in two ways. First, they bind to phosphotyrosines on cytokine receptors via an SH2 domain, thereby competing with STATs. Second, they recruit E3 ubiquitin ligases via a domain called the SOCS box. This leads to ubiquitination of the activator kinase, JAK2, which is subsequently degraded via the proteasome.
In the mammary gland, loss of SOCS1 and SOCS2 accelerated alveologenesis and rescued the phenotype of PrlR+/− mice, consistent with SOCS1 and SOCS2 exerting a negative regulatory effect downstream in the PrlR signaling pathway [50, 51].
Another negative regulator of prolactin signaling is Caveolin-1. Caveolin family members are scaffold proteins thought to organize and concentrate different lipids and lipid modified proteins within caveola membranes. It was observed that caveolin-1 is down modulated during late pregnancy and lactation as a consequence of prolactin signaling [52]. Indeed, overexpression of caveolin in HC11 cells reduced prolactin-induced activation of a β-casein reporter. Deletion of caveolin-1 led to premature STAT5 activation, alveologenesis and milk protein gene expression [53]. Caveolin-1 and -3 share conserved residues with SOCS proteins in the pseudosubstrate domain that interacts with JAK2 [53].
Deletion of JAK2 resulted in embryonic lethality. To analyze the role of its gene product in the mammary epithelium, Shillingford et al. rescued mammary anlage from JAK2−/− embryos by transplantation [36], whereas Wagner et al. used an MMTV–Cre transgene to delete the first coding exon, a floxed exon 2 [35]. In both mutant models alveologenesis and lactogenic/secretory differentiation were completely blocked, and STAT5 phosphorylation was abrogated [35, 36] consistent with JAK2 being a central mediator of PrlR signaling.
Germ line deletion of STAT5a affected the mammary gland [54], resulting in impaired alveologenesis and lactogenesis whereas inactivation of STAT5b expressed at ten-fold lower levels in the mammary gland [54], did not adversely affect the organ [55]. Ablation of both STAT5a and STAT5b strongly aggravated the phenotype [56]; STAT5 deficient mammary epithelia failed to form alveoli and to express milk protein genes [38]. Interestingly, while the PrlR deficient epithelium showed lumen formation in the smallest ducts, the STAT5 deficient epithelial ducts were not distended and, instead, had defects in cell–cell contacts [38]. This indicates that STAT5 function is not limited to mediating the effects of prolactin[57]. Indeed it can be activated by a number of receptor tyrosine kinases by Src dependent and independent mechanisms [58]. Moreover, deletion of STAT5 from the secretory epithelial cells during pregnancy resulted in cell death indicating that STAT5 function is not only required for the functional differentiation of the mammary epithelial cells, but also for their maintenance and survival [37]. Interestingly, STAT5 phosphorylation is strikingly reduced in ErbB4 deficient mammary glands, although prolactin signaling appears to be intact as assessed by STAT5 phosphorylation upon acute prolactin treatment [47]. Prolonged progesterone treatment induced STAT5 phosphorylation in the mammary epithelium of PrlR−/− but not ErbB4−/− mice, suggesting that ErbB4 is essential for STAT5 phosphorylation during late pregnancy [47]. The significance of the ErbB4 versus PrlR-induced STAT5 phosphorylation is unclear. One possibility is that HRGα-induced ErbB4 activation functions as a backup mechanism to ensure adequate STAT5 phosphorylation when PrlR signaling is down modulated via negative feedback loops. It is not known whether PrlR and ErbB4 receptors are coexpressed on the same cells and, if they are, whether they colocalize within cell membranes. Site mapping has shown that ErbB4 phosphorylates STAT5 on a residue distinct from those phosphorylated by the PrlR [47, 59], suggesting that different signaling pathways are activated by these receptors. Thus, it is conceivable that both ErbB4 and PrlR activate STAT5 but in discrete cellular compartments, leading to the activation of different downstream targets and, consequently, distinct biological outcomes. Long et al. report that ErbB4 phosphorylates STAT5 on a residue distinct from those phosphorylated by the PrlR, [47, 59] thus different protein–protein interactions could be elicited resulting in differential activation of downstream signaling pathways.
Recently, the ets transcription factor family member, ELF-5, which is widely expressed in epithelia of exocrine glands was established as a crucial mediator of prolactin signaling in the mammary gland [60]. Deletion of this transcription factor resulted in early embryonic lethality. Heterozygote females were healthy but failed to lactate. Transplantation experiments established that ELF-5 was required in the mammary epithelium for alveologenesis and milk protein expression both of which were strikingly reduced in the mammary glands of heterozygote females [61]. The levels of ELF-5 expression were reduced in PrlR+/− mammary glands, conversely, PrlR expression levels were not altered by decreased levels of ELF-5 consistent with this transcription factor being downstream of prolactin signaling [61]. Ectopic expression of ELF-5 in PrlR−/− epithelium rescued both alveologenesis and milk gene expression indicating that this transcription factor can substitute for prolactin signaling [51]. How ELF-5 relates to STAT5 remains to be elucidated. Interestingly, in T-cells STAT5 has been found to interact both physically and functionally with the ELF-5 cousins Ets-1 and Ets-2 [62].
Another transcription factor required in the mammary epithelium for both alveologenesis and lactogenic differentiation is the helix-loop-helix inhibitor ID2 [46, 63, 64]. The absence of this transcription factor does not affect the expression levels of C/EBPβ, cyclinD1, PrlR nor STAT5a or b but was shown to substantially decrease STAT5 DNA binding activity [63]. This suggests that ID2 modulates STAT5 transcriptional activity downstream of prolactin signaling. Recently, ID2 itself was shown to be downstream of RANKL, and to be required for RANKL-induced proliferation of mouse mammary epithelial cells in vitro [46]. It was found in the nucleus of wildtype cells during pregnancy, but it did not localize to nuclei in RANKL−/− mammary epithelia [46]. These findings point to the intriguing possibility that ID2 links RANKL and prolactin signaling pathways.
The CCAAT/enhancer binding proteins (C/EBP) are part of the family of leucine-zipper DNA binding (bZIP) proteins and regulate transcription by binding as homo- or heterodimers to a common nucleotide consensus site [65]. Mammary epithelium deficient for C/EBPβ fails to develop alveoli and does not undergo lactogenic differentiation [3, 66]. This phenotype is difficult to interpret because the preceding ductal morphogenesis was severely impaired resulting in a less complex ductal system and dilated ducts [66]. Moreover, expression levels of PR and PrlR were increased and the expression pattern of these receptors, as well as the response to hormonal stimulation, was altered [67] suggesting C/EBPβ is upstream of the hormonal signaling pathways that control alveologenesis. Recent work showed that C/EBPβ mediates β-casein induction by RANKL and regulates the expression of ID2 [68]. These observations indicate that several loops are built into the intracellular signaling network that controls alveologenesis and lactogenic differentiation.
Another transcription factor recently implicated in alveologenesis and lactogenic differentiation is LMO4, a member of the LIM-only family of zinc-finger proteins. Deletion of floxed alleles of this gene during pregnancy by means of a WAP–Cre transgene, partially blocks alveologenesis and milk protein synthesis, mutant dams, however, are still able to nurse their offspring [14]. How ELF5, ID2, C/EBPβ, and LMO4 interact with each other, whether they are active within the same cells and at the same promoters, at the same time or sequentially are open questions.
Finally, loss of the X-linked inhibitor of apoptosis protein (XIAP), a member of the inhibitor of apoptosis proteins family of caspase inhibitors that selectively binds and inhibits caspases-3, -7 and -9, results in impaired lactogenic differentiation and alveolar morphogenesis [13]. The mutant mammary glands showed delayed STAT5 phosphorylation and NFκB activity.
Cyclin D1 and the GR are the only intracellular players identified to date that selectively affect alveolar morphogenesis and not lactogenic differentiation [69]. Based on the well-established role of D-type cyclins in G1 progression, the failure of cyclin D1 deficient mice to undergo alveologenesis was attributed to a defect in proliferation during pregnancy[41, 70]. Recent findings that the mammary defect of cyclin D1 deficient mice was rescued when the gene is replaced by a cyclin D1 mutant that does not interact with the cyclin dependent kinases 4 and 6 (CDK4 and CDK6) and hence cannot promote cell cycle progression [71] indicate that cyclin D1 has a role beyond its well-established cell cycle regulatory function. Indeed, cyclin D1 has previously been shown to affect ER mediated transcription albeit in overexpression studies [72, 73]. Unraveling the new interactors and mediators of protein protein interactions, cyclin D1 function in the mammary epithelium should provide insights into the mechanisms underlying alveologenesis.
Taken together a signaling network that emerges controls alveologenesis and lactogenic differentiation in which prolactin signaling is the central hub (Fig. 2).
Cell Adhesion
Over the past decade the importance of epithelial polarity and adhesion for secretory function/differentiation has become apparent. The interaction of mammary epithelial cells with the extracellular matrix is mediated by integrins. While mammary glands developed normally in the absence of α3, or α6 integrins [74] deletion of β1-intgrin from the mammary epithelium during pregnancy, resulted in reduced alveologenesis, disrupted alveolar structure and reduced expression of the milk protein β-casein [75, 76].
Similarly, deletion of α-catenin using a Cre transgene under the control of either an MMTV or a WAP promoter, resulted in a lactation defect. The mutant mothers developed alveolar structures that failed to expand. The luminal cells showed defects in adhesion and loss of epithelial cell polarity, and reduced milk protein gene expression. The phenotype was more pronounced in MMTV–Cre deleted than WAP–Cre deleted mutants [77]. Similarly, deletion of E-cadherin by an MMTV–Cre transgene specifically affects terminal differentiation. The phenotype became apparent during pregnancy, because alveoli failed to expand in the knockout mice, and histological sections revealed no fat droplets. Further analysis confirmed this observation by demonstrating that the milk protein genes, β-casein and WAP, were expressed in the knockout at levels much lower compared to wild type control and mothers were unable to feed their pups [7].
Taken together, these observations indicate that intact cell adhesion is a prerequisite for successful alveologenesis and lactogenic differentiation. Why does the deletion of such important adhesion molecules as E-cadherin or α-catenin, have no obvious effect on ductal morphogenesis but is clearly detrimental for alveologenesis and lactogenic differentiation? A simple explanation might be that the deletion efficiency is higher during pregnancy when the MMTV promoter shows maximum activity than in the virgin mouse. Alternatively, compensatory mechanisms might be available that help the mammary epithelium to adjust to the challenges of ductal morphogenesis, such as the SLITs/Netrins [78]. Ultimately, these backup mechanisms fail when the epithelium needs to withstand the pressure generated by the secretions that accumulate in the ducts as the cells differentiate.
Outlook
Over the past 15 years, mouse genetics have revealed many of the components of the signaling network that controls alveologenesis and lactogenic differentiation (as illustrated in Fig. 2). Further factors are likely to be identified and new interactions between the signaling cascades and cellular functions should be revealed.
There is a clear need for more conditional/inducible mutant models. As has been illustrated by the comparison of the germ line and conditional deletions of LMO4 and XIAP, the mammary epithelium is fully capable of adapting to the lack of many different genes during development. It is conceivable that novel functions of PR signaling and C/EBPβ during alveologenesis and lactogenic differentiation will be uncovered by deleting these genes specifically during late stages of mammary gland development. Pursuing these questions will require new powerful inducible models [79].
This review of the mammary gland has focused almost exclusively on the targets of the prolactin signaling in the luminal epithelial cells. Currently, we know little about the physiological function of the myoepithelial cells and the changes in the stroma required for alveologenesis and lactogenic differentiation (for review, see July 2005 issue of this journal). Evidence has accumulated that the myoepithelium is more than an innocent bystander during breast carcinogenesis [80] and the same seems to apply to development [81]. Further insights will likely come from increasingly complex 3D culture studies [82].
With regards to the stromal contribution new models are needed. Many “stroma-specific” promoters express not only in the mammary gland but are active in the connective tissue of numerous organs, this limits their use in driving Cre. Furthermore, we will need to refine our appreciation of the stroma. Obviously, the stromal fibroblasts in contact with the basal lamina have very different role from the fat cells. Cell-type specific promoters used to drive Cre transgenes will be instrumental to gaining new insights.
Capillary plexuses invest the secretory alveoli. How do these develop? How is the establishment of new blood vessels coordinated with epithelial outgrowth? Lymph flow from the lactating gland is high, how is the lymph flow established? Ever more powerful imaging techniques provide new dynamic views of mammary gland development in vivo that may provide answers to these questions.
Abbreviations
- WAP:
-
whey acidic protein
- XIAP:
-
x-linked inhibitor of apoptosis protein
- GH:
-
growth hormone
- PTHrP:
-
parathyroid hormone related peptide
- GR:
-
glucocorticoid receptor
- ER:
-
estrogen receptor
- PR:
-
progesterone receptor
- PrlR:
-
prolactin receptor
- RANKL:
-
receptor activator of NFκB ligand
- EGF:
-
epidermal growth factor
- HRG1:
-
heregulin 1
- GAS:
-
γ-interferon activation sequence
- JAK2:
-
janus 2 kinase
- SOCS:
-
suppressor of cytokine signaling proteins
- STAT:
-
signal transducer and activator of transcription
- C/EBP:
-
CCAAT/enhancer binding proteins
- CDK:
-
cyclin dependent kinase
- MMTV:
-
mouse mammary tumor virus
- LMO4:
-
LIM domain only 4
- ErbB4:
-
v-erb-a erythroblastic leukemia viral oncogene homolog 4
- ID2:
-
inhibitor of DNA binding 2
- Ets:
-
v-ets erythroblastosis virus E26 oncogene homolog 1
- ELF-5:
-
E74-like factor 5
- IGF-2:
-
insulin-like growth factor 2
- ERK1:
-
extracellular-signal-regulated kinase 1
- IKKα:
-
Inhibitor of kappa B kinase alpha
References
Brisken C. Hormonal control of alveolar development and its implications for breast carcinogenesis. J Mammary Gland Biol Neoplasia 2002;7:39–48.
Neville MC, McFadden TB, Forsyth I. Hormonal regulation of mammary differentiation and milk secretion. J Mammary Gland Biol Neoplasia 2002;7:49–66.
Robinson GW, Johnson PF, Hennighausen L, Sterneck E. The C/EBPbeta transcription factor regulates epithelial cell proliferation and differentiation in the mammary gland. Genes Dev 1998;12:1907–16.
Jensen EV, Cheng G, Palmieri C, Saji S, Makela S, Van Noorden S, et al. Estrogen receptors and proliferation markers in primary and recurrent breast cancer. Proc Natl Acad Sci USA 2001;98:15197–202.
Neville MC, Morton J. Physiology and endocrine changes underlying human lactogenesis II. J Nutr 2001;131:3005S–8S.
MacMahon B, Cole P, Lin TM, Lowe CR, Mirra AP, Ravnihar B, et al. Age at first birth and breast cancer risk. Bull World Health Organ 1970;43:209–21.
Boussadia O, Kutsch S, Hierholzer A, Delmas V, Kemler R. E-cadherin is a survival factor for the lactating mouse mammary gland. Mech Dev 2002;115:53–62.
Hennighausen L, Robinson GW. Information networks in the mammary gland. Nat Rev Mol Cell Biol 2005;6:715–25.
Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 2005;6:507–12.
Brisken C, Park S, Vass T, Lydon JP, O’Malley BW, Weinberg RA. A paracrine role for the epithelial progesterone receptor in mammary gland development. Proc Natl Acad Sci USA 1998;95:5076–81.
Robinson GW, Hennighausen L, Johnson PF. Side-branching in the mammary gland: the progesterone-Wnt connection. Genes Dev 2000;14:889–94.
Wintermantel TM, Mayer AK, Schutz G, Greiner EF. Targeting mammary epithelial cells using a bacterial artificial chromosome. Genesis 2002;33:125–30.
Olayioye MA, Kaufmann H, Pakusch M, Vaux DL, Lindeman GJ, Visvader JE. XIAP-deficiency leads to delayed lobuloalveolar development in the mammary gland. Cell Death Differ 2005;12:87–90.
Sum EY, Shackleton M, Hahm K, Thomas RM, O’Reilly LA, Wagner KU, et al. Loss of the LIM domain protein Lmo4 in the mammary gland during pregnancy impedes lobuloalveolar development. Oncogene 2005;24:4820–8.
Clevenger CV, Plank TL. Prolactin as an autocrine/paracrine factor in breast tissue. J Mammary Gland Biol Neoplasia 1997;2:59–68.
Mol JA, Lantinga-van Leeuwen I, van Garderen E, Rijnberk A. Progestin-induced mammary growth hormone (GH) production. Adv Exp Med Biol 2000;480:71–6.
Woodside B, Abizaid A, Walker C. Changes in leptin levels during lactation: implications for lactational hyperphagia and anovulation. Horm Behav 2000;37:353–65.
Lippuner K, Zehnder HJ, Casez JP, Takkinen R, Jaeger P. PTH-related protein is released into the mother’s bloodstream during location: evidence for beneficial effects on maternal calcium–phosphate metabolism. J Bone Miner Res 1996;11:1394–9.
Naylor MJ, Oakes SR, Gardiner-Garden M, Harris J, Blazek K, Ho TW, et al. Transcriptional changes underlying the secretory activation phase of mammary gland development. Mol Endocrinol 2005;19:1868–83.
Rosen JM, Zahnow C, Kazansky A, Raught B. Composite response elements mediate hormonal and developmental regulation of milk protein gene expression. Biochem Soc Symp 1998;63:101–13.
Wintermantel TM, Bock D, Fleig V, Greiner EF, Schutz G. The epithelial glucocorticoid receptor is required for the normal timing of cell proliferation during mammary lobuloalveolar development but is dispensable for milk production. Mol Endocrinol 2005;19:340–9.
Capuco AV, Kahl S, Jack LJ, Bishop JO, Wallace H. Prolactin and growth hormone stimulation of lactation in mice requires thyroid hormones. Proc Soc Exp Biol Med 1999;221:345–51.
Tsai MJ, O’Malley BW. Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 1994;63:451–86.
Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell 1995;83:851–7.
Jarvinen TA, Pelto-Huikko M, Holli K, Isola J. Estrogen receptor beta is coexpressed with ERalpha and PR and associated with nodal status, grade, and proliferation rate in breast cancer. Am J Pathol 2000;156:29–35.
Pelletier G, El-Alfy M. Immunocytochemical localization of estrogen receptors alpha and beta in the human reproductive organs. J Clin Endocrinol Metab 2000;85:4835–40.
Saji S, Jensen EV, Nilsson S, Rylander T, Warner M, Gustafsson JA. Estrogen receptors alpha and beta in the rodent mammary gland. Proc Natl Acad Sci USA 2000;97:337–42.
Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors alpha (ERalpha) and beta (ERbeta) on mouse reproductive phenotypes. Development 2000;127:4277–91.
Mallepell S, Krust A, Chambon P, Brisken C. Paracrine signaling through the epithelial estrogen receptor alpha is required for proliferation and morphogenesis in the mammary gland. Proc Natl Acad Sci USA 2006;103:2196–201.
Forster C, Makela S, Warri A, Kietz S, Becker D, Hultenby K, et al. Involvement of estrogen receptor beta in terminal differentiation of mammary gland epithelium. Proc Natl Acad Sci USA 2002;99:15578–83.
Lydon JP, DeMayo FJ, Funk CR, Mani SK, Hughes AR, Montgomery CA, Jr., et al. Mice lacking progesterone receptor exhibit pleiotropic reproductive abnormalities. Genes Dev 1995;9:2266–78.
Mulac-Jericevic B, Lydon JP, DeMayo FJ, Conneely OM. Defective mammary gland morphogenesis in mice lacking the progesterone receptor B isoform. Proc Natl Acad Sci USA 2003;100:9744–9.
Brisken C, Kaur S, Chavarria TE, Binart N, Sutherland RL, Weinberg RA, et al. Prolactin controls mammary gland development via direct and indirect mechanisms. Dev Biol 1999;210:96–106.
Ormandy CJ, Camus A, Barra J, Damotte D, Lucas B, Buteau H, et al. Null mutation of the prolactin receptor gene produces multiple reproductive defects in the mouse. Genes Dev 1997;11:167–78.
Wagner KU, Krempler A, Triplett AA, Qi Y, George NM, Zhu J, et al. Impaired alveologenesis and maintenance of secretory mammary epithelial cells in Jak2 conditional knockout mice. Mol Cell Biol 2004;24:5510–20.
Shillingford JM, Miyoshi K, Robinson GW, Grimm SL, Rosen JM, Neubauer H, et al. Jak2 is an essential tyrosine kinase involved in pregnancy-mediated development of mammary secretory epithelium. Mol Endocrinol 2002;16:563–70.
Cui Y, Riedlinger G, Miyoshi K, Tang W, Li C, Deng CX, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol 2004;24:8037–47.
Miyoshi K, Shillingford JM, Smith GH, Grimm SL, Wagner KU, Oka T, et al. Signal transducer and activator of transcription (Stat) 5 controls the proliferation and differentiation of mammary alveolar epithelium. J Cell Biol 2001;155:531–42.
Galsgaard ED, Friedrichsen BN, Nielsen JH, Moldrup A. Expression of dominant-negative STAT5 inhibits growth hormone- and prolactin-induced proliferation of insulin-producing cells. Diabetes 2001;50(Suppl 1):S40–1.
Ma FY, Anderson GM, Gunn TD, Goffin V, Grattan DR, Bunn SJ. Prolactin specifically activates signal transducer and activator of transcription 5b in neuroendocrine dopaminergic neurons. Endocrinology 2005;146:5112–9.
Brisken C, Ayyannan A, Nguyen C, Heineman A, Reinhardt F, Tan J, et al. IGF-2 is a mediator of prolactin-induced morphogenesis in the breast. Dev Cell 2002;3:877–87.
Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell 2000;103:41–50.
Srivastava S, Matsuda M, Hou Z, Bailey JP, Kitazawa R, Herbst MP, et al. Receptor activator of NF-kappaB ligand induction via Jak2 and Stat5a in mammary epithelial cells. J Biol Chem 2003;278:46171–8.
Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, et al. IKKalpha provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 2001;107:763–75.
Jones DH, Nakashima T, Sanchez OH, Kozieradzki I, Komarova SV, Sarosi I, et al. Regulation of cancer cell migration and bone metastasis by RANKL. Nature 2006;440:692–6.
Kim NS, Kim HJ, Koo BK, Kwon MC, Kim YW, Cho Y, et al. Receptor activator of NF-kappaB ligand regulates the proliferation of mammary epithelial cells via Id2. Mol Cell Biol 2006; 26:1002–13.
Long W, Wagner KU, Lloyd KC, Binart N, Shillingford JM, Hennighausen L, et al. Impaired differentiation and lactational failure of Erbb4-deficient mammary glands identify ERBB4 as an obligate mediator of STAT5. Development 2003;130:5257–68.
Jones FE, Welte T, Fu XY, Stern DF. ErbB4 signaling in the mammary gland is required for lobuloalveolar development and Stat5 activation during lactation. J Cell Biol 1999;147:77–88.
Watson CJ, Burdon TG. Prolactin signal transduction mechanisms in the mammary gland: the role of the Jak/Stat pathway. Rev Reprod 1996;1:1–5.
Lindeman GJ, Wittlin S, Lada H, Naylor MJ, Santamaria M, Zhang JG, Blazek KD, Kazlaukas M, Hilton HN, Wittlin S, Alexander WS, Lindeman GJ. Visvader JE, Ormandy CJ. SOCS1 deficiency results in accelerated mammary gland development and rescues lactation in prolactin receptor-deficient mice. Genes Dev 2001;15:1631–6.
Harris J, Stanford PM, Sutherland K, Oakes SR, Naylor MJ, Robertson FG, Blazek KD, Kazlaukas M, Hilton HN, Wittlin S, Alexandra WS, Lindeman GJ, Visvader JE, Ormandy CJ. Socs2 and Elf5 mediate prolactin-induced mammary gland development. Mol Endocrinol 2006 May; 20(5):1177–87.
Park DS, Lee H, Riedel C, Hulit J, Scherer PE, Pestell RG, et al. Prolactin negatively regulates caveolin-1 gene expression in the mammary gland during lactation, via a Ras-dependent mechanism. J Biol Chem 2001;276:48389–97.
Park DS, Lee H, Frank PG, Razani B, Nguyen AV, Parlow AF, et al. Caveolin-1-deficient mice show accelerated mammary gland development during pregnancy, premature lactation, and hyperactivation of the Jak-2/STAT5a signaling cascade. Mol Biol Cell 2002;13:3416–30.
Liu X, Robinson GW, Wagner KU, Garrett L, Wynshaw-Boris A, Hennighausen L. Stat5a is mandatory for adult mammary gland development and lactogenesis. Genes Dev 1997;11:179–86.
Udy GB, Towers RP, Snell RG, Wilkins RJ, Park SH, Ram PA, et al. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc Natl Acad Sci USA 1997;94:7239–44.
Teglund S, McKay C, Schuetz E, van Deursen JM, Stravopodis D, Wang D, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell 1998;93:841–50.
Brisken C, Ayyanan A, Doppler W. Prolactin signaling and Stat5: going their own separate ways? Breast Cancer Res 2002;4:209–12.
Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev 2004;15:435–55.
Clark DE, Williams CC, Duplessis TT, Moring KL, Notwick AR, Long W, et al. ERBB4/HER4 potentiates STAT5A transcriptional activity by regulating novel STAT5A serine phosphorylation events. J Biol Chem 2005;280:24175–80.
Lapinskas EJ, Palmer J, Ricardo S, Hertzog PJ, Hammacher A, Pritchard MA. A major site of expression of the ets transcription factor Elf5 is epithelia of exocrine glands. Histochem Cell Biol 2004;122:521–6.
Zhou J, Chehab R, Tkalcevic J, Naylor MJ, Harris J, Wilson TJ, et al. Elf5 is essential for early embryogenesis and mammary gland development during pregnancy and lactation. Embo J 2005;24:635–44.
Rameil P, Lecine P, Ghysdael J, Gouilleux F, Kahn-Perles B, Imbert J. IL-2 and long-term T cell activation induce physical and functional interaction between STAT5 and ETS transcription factors in human T cells. Oncogene 2000;19:2086–97.
Mori S, Nishikawa SI, Yokota Y. Lactation defect in mice lacking the helix-loop-helix inhibitor Id2. Embo J 2000;19:5772–81.
Miyoshi K, Meyer B, Gruss P, Cui Y, Renou JP, Morgan FV, et al. Mammary epithelial cells are not able to undergo pregnancy-dependent differentiation in the absence of the helix-loop-helix inhibitor Id2. Mol Endocrinol 2002;16:2892–901.
Williams SC, Cantwell CA, Johnson PF. A family of C/EBP-related proteins capable of forming covalently linked leucine zipper dimers in vitro. Genes Dev 1991;5:1553–67.
Seagroves TN, Krnacik S, Raught B, Gay J, Burgess-Beusse B, Darlington GJ, et al. C/EBPbeta, but not C/EBPalpha, is essential for ductal morphogenesis, lobuloalveolar proliferation, and functional differentiation in the mouse mammary gland. Genes Dev 1998;12:1917–28.
Grimm SL, Seagroves TN, Kabotyanski EB, Hovey RC, Vonderhaar BK, Lydon JP, et al. Disruption of steroid and prolactin receptor patterning in the mammary gland correlates with a block in lobuloalveolar development. Mol Endocrinol 2002;16:2675–91.
Karaya K, Mori S, Kimoto H, Shima Y, Tsuji Y, Kurooka H, et al. Regulation of Id2 expression by CCAAT/enhancer binding protein beta. Nucleic Acids Res 2005;33:1924–34.
Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev 1995;9:2364–72.
Sicinski P, Donaher JL, Parker SB, Li T, Fazeli A, Gardner H, et al. Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 1995;82:621–30.
Landis MW, Pawlyk BS, Li T, Sicinski P, Hinds PW. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cells 2006;9:13–22.
Zwijsen RM, Wientjens E, Klompmaker R, van der Sman J, Bernards R, Michalides RJ. CDK-independent activation of estrogen receptor by cyclin D1. Cell 1997;88:405–15.
Ewen ME, Lamb J. The activities of cyclin D1 that drive tumorigenesis. Trends Mol Med 2004;10:158–62.
Klinowska TC, Alexander CM, Georges-Labouesse E, Van der Neut R, Kreidberg JA, Jones CJ, et al. Epithelial development and differentiation in the mammary gland is not dependent on alpha 3 or alpha 6 integrin subunits. Dev Biol 2001;233:449–67.
Li N, Zhang Y, Naylor MJ, Schatzmann F, Maurer F, Wintermantel T, et al. Beta1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. Embo J 2005;24:1942–53.
Naylor MJ, Li N, Cheung J, Lowe ET, Lambert E, Marlow R, et al. Ablation of beta1 integrin in mammary epithelium reveals a key role for integrin in glandular morphogenesis and differentiation. J Cell Biol 2005;171:717–28.
Nemade RV, Bierie B, Nozawa M, Bry C, Smith GH, Vasioukhin V, et al. Biogenesis and function of mouse mammary epithelium depends on the presence of functional alpha-catenin. Mech Dev 2004;121:91–9.
Strickland P, Shin GC, Plump A, Tessier-Lavigne M, Hinck L. Slit2 and netrin 1 act synergistically as adhesive cues to generate tubular bi-layers during ductal morphogenesis. Development 2006;133:823–32.
Gunther EJ, Belka GK, Wertheim GB, Wang J, Hartman JL, Boxer RB, et al. A novel doxycycline-inducible system for the transgenic analysis of mammary gland biology. Faseb J 2002;16:283–92.
Adriance MC, Inman JL, Petersen OW, Bissell MJ. Myoepithelial cells: good fences make good neighbors. Breast Cancer Res 2005;7:190–7.
Teuliere J, Faraldo MM, Deugnier MA, Shtutman M, Ben-Ze’ev A, Thiery JP, et al. Targeted activation of beta-catenin signaling in basal mammary epithelial cells affects mammary development and leads to hyperplasia. Development 2005;132:267–77.
Nelson CM, Bissell MJ. Modeling dynamic reciprocity: engineering three-dimensional culture models of breast architecture, function, and neoplastic transformation. Semin Cancer Biol 2005;15:342–52.
Acknowledgments
We thank Manfred Beleut for carefully reading the manuscript. This work was supported by funds from the National Center of Competence in Research in Molecular Oncology and Oncosuisse.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Brisken, C., Rajaram, R.D. Alveolar and Lactogenic Differentiation. J Mammary Gland Biol Neoplasia 11, 239–248 (2006). https://doi.org/10.1007/s10911-006-9026-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-006-9026-0