
Abstract
Polycondensation of boric acid and vinyltriethoxysilane in 1:2, 1:1.5 and 1:1 mole ratio in diglyme at 83–87 °C for 3 h using hydrochloric acid as catalyst afforded vinylfunctionalized borosiloxane oligomers soluble in the reaction medium. Complete removal of ethanol, the by-product, and diglyme rendered the oligomers intractable due to the advancement of polycondensation. They were characterized by FTIR and TGA and converted to ceramics by heat treatment at 900 °C, 1500 °C and 1650 °C in argon atmosphere. The ceramics obtained were characterized by IR, Raman, 13C-and 29Si-solid state NMR spectroscopy and XRD. These studies infer the formation of SiOC/SiBOC glass on pyrolysis of these oligomers at 900 °C and onset of formation of β-SiC at 1500 °C. On further heat treatment at 1650 °C, complete conversion of the ceramic to a mixture of α and β-SiC was observed along with the presence of diamond like carbon phases.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
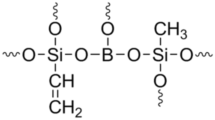
Polymer-derived Ceramics (PDCs) have been the target by researchers for more than four decades in order to obtain new ceramic systems with improved high temperature properties, oxidation resistance and corrosion resistance [1, 2]. Synthesis of a large number of Si-based polymeric systems such as polysilanes, polycarbosilanes, polysilazanes, polycarbosilazanes and polysilylcarbodiimides and their conversion to non-oxide ceramics such as SiC, Si3N4 and SiCN have been reported [3,4,5,6,7]. In recent years, polysiloxanes [8,9,10,11,12], polysilsesquioxanes [13,14,15,16] and polyborosiloxanes [17,18,19,20,21,22,23,24,25] have been explored extensively in view of their ease of synthesis, low cost and the ability to give SiOC and SiBOC amorphous ceramics which can only be formed by PDC route. The inclusion of boron in SiOC results in the improvement of thermooxidative stability besides influencing the crystallization kinetics of SiOC [17, 26, 27]. In view of this, polyborosiloxanes have gained significance as matrix resins for Ceramic Matrix Composites (CMCs) [28,29,30,31,32,33,34,35], binder for making ceramic bodies [36] and as precursors for SiBOC/SiC coating [37], SiBOC fibre [38,39,40] and SiBOC foam [41].
Polyborosiloxanes are prepared usually by sol–gel process by reacting boric acid or its esters with alkoxysilanes/acetoxysilanes/organoalkoxysilanes in aqueous medium [42,43,44,45]. However, over the years, polyborosiloxanes have been prepared under non-aqueous conditions by reacting boric acid/phenylboronic acid with organoalkoxysilanes in diglyme or dioxane medium [21, 46,47,48,49] or directly [18,19,20, 48,49,50]. The advantage of the non-aqueous process is that the formation of Si–O–B bond is ensured and hydrolytic cleavage of Si–O–B bond already formed is eliminated [51]. Following our initial success on the synthesis of resinous polyborosiloxane from phenyltrimethoxysilane and boric acid [46,47,48], in our laboratory, attention has been focused on the synthesis of polyborosiloxanes from a variety of organoalkoxysilane and boric acid/phenylboronic acid by non-aqueous sol–gel process using different monomer feed ratios and evolution of polyborosiloxane-derived SiBOC/SiC ceramics [19,20,21,22]. We have observed that the extent of conversion of SiOC/SiBOC to β-SiC with respect to heat treatment temperature is controlled by exchange reaction of SiOC/SiBOC units and carbothermic reduction involving SiOC/SiBOC and free carbon present in SiOC/SiBOC glass matrix [19, 20, 22]. Understanding the microstructure of ceramics formed from polyborosiloxanes at different heat treatment temperatures is of significance particularly when these precursors are to be used as precursor matrix for CMCs. Poly(vinylborosiloxane) an important precursor for SiBOC/SiC as it gives maximum ceramic residue among the polyborosiloxanes. From our laboratory we reported the synthesis of poly(vinylborosiloxane) by reacting boric acid with vinyltriethoxysilane (VTEOS) or vinyltris(2-methoxyethoxy)silane [VTMEOS] by non-aqueous sol–gel process in diglyme. Due to the lower reactivity of VTMEOS, poly(vinylborosiloxane) obtained is soluble even after the removal of ethanol formed as by-product and solvent diglyme. Under this processing condition, poly(vinylborosiloxane) formed from VTEOS got gelled. In view of the processability of poly(vinylborosiloxane) synthesized from VTMEOS, we have studied the conversion of poly(vinylborosiloxane), synthesized from boric acid and VTMEOS using three different monomer feed ratios, into ceramics by heat treatment at different temperatures. Our earlier studies indicated that nature of organic substituent such as phenyl, methyl, vinyl and allyl on silicon has a direct bearing on the conversion of SiBOC ceramic to SiC ceramic [20,21,22, 48,49,50]. Thus, it would be interest to study the influence of alkoxy group attached to Si on the ceramic conversion of poly(borosiloxane)s synthesized using such alkoxysilanes. In the present study, we focus our attention on the synthesis of poly(vinylborosiloxane) by non-aqueous sol–gel process by using VTEOS in place of VTMEOS, and transformation and microstructure evolution of SiBOC ceramics formed by pyrolysis of this precursor.
2 Experimental
2.1 Synthesis of Poly(vinylborosiloxane)s
Polyvinylborosiloxane was synthesized by reacting boric acid (BA) with vinyltriethoxysilane (VTEOS) in diglyme at 83–87 °C using hydrochloric acid as the catalyst following the procedure described in our earlier publication [21]. Though the vinyl-functionalized borosiloxane oligomers formed were soluble in the reaction medium, removal of ethanol, the by-product, by flash evaporation, rendered them insoluble in the reaction medium. At this stage, diglyme was removed by distillation under reduced pressure. The sample was dried in a vacuum oven at 100 °C for 5 h. Polyvinylborosiloxanes were synthesized using different monomer feed ratios (BA:VTEOS) 1:2, 1:1.5 and 1:1 were designated as BSiV-1, BSiV-2 and BSiV-3 respectively.
2.2 Polymer to Ceramic Transformation
Poly(vinylborosiloxane)s were heat treated at 900 °C, 1500 °C and 1650 °C in an inconel furnace, an alumina tubular furnace and a graphite furnace respectively under the flow of argon (~ 50 ml/min) to obtain ceramics following the procedure described elsewhere [21].
2.3 Characterization
FTIR spectra of the oligomers and the ceramics obtained after heat treatment at different temperatures were recorded on a Perkin Elmer GX spectrometer. Thermogravimetric (TG) analysis of the borosiloxane oligomers was performed on a TA Instruments SDT 2960 under nitrogen atmosphere at a heating rate of 10 °C/min from room temperature to 900 °C. Raman spectral characterization of the ceramics were done in WITec alpha 300R Confocal Raman microscope equipped with a 100X air objective and 600 grooves/mm grating. 532 nm laser source was used for excitation and five spectra were accumulated with an integration time of 1 sec to obtain a single spectrum. X-ray diffraction (XRD) patterns of ceramic powder samples were recorded on Xpert-Pro MPD instrument with Cu Kα (1.54 Å). Boron content of the ceramics obtained was determined by wet analysis [52].
29Si-Magic Angle Spinning (MAS) NMR and 13C-MAS NMR were acquired at 79.5 MHz and 100.6 MHz respectively using Bruker Avance III HD NMR spectrometer (9.1 T) equipped with 4 mm MAS probe. 5 kHz spinning rate and conventional one pulse experiment were used for both the nuclei. For 29Si-NMR pulse delay of 500 s with 45° excitation pulse was used to take 12 scans, whereas for 13C NMR pulse delay of 5 s with 90° excitation pulse was used to take 13 k scans. Both the 29Si- and 13C-NMR spectra were Fourier transformed with 100 Hz of line broadening.
3 Results and Discussion
3.1 Synthesis of Poly(vinylborosiloxane)s
Poly(vinylborosiloxane)s were synthesized from boric acid and VTEOS as shown in Scheme 1. Formation of Si–O–B linkages take place due to the condensation of B–OH group of boric acid and Si–OEt group of VTEOS resulting in the elimination of ethanol. It is noticed that for all the monomer feed ratios, the oligomers formed are soluble in the reaction medium. While carrying out flash evaporation to remove the solvent (diglyme) and the by-product (ethanol), viscosity of the solution gradually increased and finally, a gel was obtained which on further drying resulted in the formation of a flaky material. The cross linked resins, thus obtained, are insoluble in solvents like acetone, chloroform, tetrahydrofuran, diglyme, toluene and dioxane. They were characterized by IR and TGA. Though complete removal of ethanol renders poly(vinylborosiloxane) insoluble, it is possible to obtain diglyme solution of poly(vinylborosiloxane) useful for applications as adhesives, coatings and matrix resin for CMCs by regulating the amount of ethanol distilled out of the reaction mixture.

Synthesis of poly(vinylborosiloxane) from boric acid (BA) and VTEOS (BA:VTEOS-1:2, BSiV-1; 1:1.5, BSiV-2; 1:1, BSiV-3)
The IR spectra of the oligomers (Fig. 1) show a peak at 3224 cm−1 corresponding to O–H stretching of B-OH groups, suggesting the presence of unreacted B-OH groups in the oligomeric structure. This is again validated by the peak at 1191 cm−1indicating B–OH deformation which is more prominent in BSiV-3, prepared using the feed ratio (1:1). The peak at 1603 cm−1 corresponding to vinyl group is observed in the IR spectrum of all the oligomers indicating the presence of vinyl group which is more prominent for BSiV-1. All the three oligomers show peak at 884 cm−1 in the IR spectra which confirms the presence of Si–O–B bonds [38].

IR spectra of poly(vinylborosiloxane): a BSiV-1, b BSiV-2 and c BSiV-3
The TG curves of BSiV-1, BSiV-2 and BSiV-3 are compared in Fig. 2. Considerable mass loss of 4.7, 9.9 and 17.2% is observed for BSiV-1, BSiV-2 and BSiV-3, respectively in the initial stage up to 170 °C and this mass loss is mainly due to the loss of water and ethanol formed from the reaction of unreacted Si–OH, B–OH and Si–OEt moieties [53]. Loss of low molecular weight oligomers/cage and cyclic polysiloxanes is responsible for the mass loss of 2, 3 and 7% for BSSiV-1, BSiV-2 and BSiV-3 respectively in the temperature range from 170 °C to 500 °C [54]. Mass loss of 4, 5 and 6% for BSiV-1, BSiV-2 and BSiV-3 respectively is recorded in the region 500–680 °C which is caused by the loss of organic moieties. It is worth pointing out that the mass loss due to the loss of organic moiety is considerably less (by 11–14%) and the ceramic residue is significantly higher (by 8–17%) for poly(vinylborosiloxane)s compared to poly(phenylborosiloxane)s [21] which is due to thermal polymerization of Si-vinyl groups resulting in crosslinking. Comparison of ceramic residue at 900 °C of VTEOS- and VTMEOS-based [22] systems indicates that the ceramic residue is higher for VTEOS-based system than VTMEOS-based system (73–90% versus 72–81%) and this has been attributed to the higher reactivity of VTEOS compared to that of VTMEOS. In the case of VTMEOS-based system [22] due to the lower reactivity of VTMEOS, the extent of branching would be less and also there would be unreacted methoxyethoxy groups. The loss of unreacted methoxyethoxy group (having higher molar mass than ethoxy) combined with lesser extent of branching would contribute to the lower ceramic residue of VTEMOS-based poly(borosiloxane).

TG curves of poly(vinylborosiloxane)s (heating rate: 10 °C/min)
3.2 Heat Treatment of Poly(vinylborosiloxane)s
The XRD patterns (Fig. 3) suggest that BSiV samples pyrolyzed at 900 °C are fully amorphous. The diffraction halo centered at 2θ = 25° corresponds to SiOC/SiBOC glass [55]. Thus, pyrolysis of the samples at 900 °C renders poly(vinylborosiloxane)s into boron modified SiOC phase where SiCO3, SiC2O2, SiC3O, BC2O and BCO2 units may be present. In the case of the pyrolyzed BSiV-3, diffraction line is observed at 28° corresponding to B2O3 which is absent in the pyrolyzed samples of BSiV-1 and BSiV-2. When the pyrolyzed samples are heat treated at 1500 °C onset of nano crystallites of β-SiC is noticed. The transformation of amorphous SiOC/SiBOC glass formed from BSiVs to β-SiC is explained by adopting the nucleation and the crystal growth mechanism of formation of β-SiC crystallites from SiOC [56]. Apart from the diffractions corresponding to β-SiC and SiBOC glass, the samples show the presence of crystalline B2O3 (2θ = 28°) and silica (2θ = 26°) depending on the monomer feed ratio (BA:VTEOS) used for the synthesis of BSiVs. The samples heat treated at 1650 °C show sharp diffraction lines at 2θ = 36, 61 and 73° which corresponds to 111, 220 and 311 crystallographic planes respectively of β-SiC and a small shoulder peak at 33° due to either traces of α-SiC or to the stacking faults in β-SiC which would have occurred during the ceramization process [57]. Though the exchange mechanism is more operative, the carbothermic reduction involving nanodomains of SiO2 rich and carbon rich phases can also take place [58]. Such a reaction is expected to be more predominant when the heat treatment temperature is further increased resulting in increased weight loss. This is clearly seen from the weight loss data of poly(vinylborosiloxane)s heat treated at 900 °C, 1500 °C and 1650 °C (Table 1). The weight loss at 1650 °C is almost the same for all the three systems indicating that the final quantity of SiC formed at 1650 °C is independent of the monomer feed ratio. However, it is seen from Table 1 that weight loss of the ceramic on increasing the heat treatment temperature from 1500 to 1650 °C is more than double for BSiV-1 and BSiV-2 (34% and 28% respectively) compared to that of BSiV-3 (12%). This is understood in view of the higher concentration of vinylsiloxy units in BSiV-1 and BSiV-2 than in BSiV-3. This would result in more pronounced carbothermic reduction involving SiO2 nanodomains and carbon nanodomains in BSiV-1 and BSiV-2 accounting for higher weight loss compared to that of BSiV-1 when the heat treatment temperature is increased from 1500 to 1650 °C. Under the experimental conditions, the formation of boron carbide (B4C) due to the carbothermic reduction of B–O units, particularly when the heat treatment temperature is increased from 1500 to 1650 °C, cannot be ruled out.

XRD patterns of ceramics from a BSiV-1 b BSiV-2 and c BSiV-3
It is noticed that the crystallite size of β-SiC calculated using Scherrer equation [59] is 13 nm for BSiV-1, 28 nm for BSiV-2 and 38 nm for BSiV-3. Boron content in BSiV precursors is expected to increase with the decrease in VTEOS concentration in the monomer feed. In line with the reports by Soraru et al. [55], this observation may be attributed to the reduction in viscosity of the SiBOC glass with increase in boron incorporation into the SiBOC network. This facilitates the mobility of carbon and silicon atoms in the glass matrix, thereby increasing the β-SiC formation. Elemental analysis indicates that boron is present in the ceramics obtained at 1650 °C (2.6 wt% for BSiV-1 heat-treated at 1650 °C) and probably it is present in the residual glassy phase as BO3 trigonal units [55, 56] bonded to silicon via Si–O–B bonds as evidenced from FTIR (Fig. 4). Thus, the ceramics obtained at 1650 °C from poly(vinylborosiloxane)s consist of β-SiC nanocrystals along with α-SiC for all the three systems.

FTIR spectra of ceramics from: a BSiV-1, b BSiV-2 and c BSiV-3
Based on our investigation on the ceramic conversion of borosiloxanes synthesized from different alkoxysilanes, we observe that both the organic substituent on silicon and the alkoxy group influence the crystallization of ceramic formed by heat treatment. As discussed earlier, poly(vinylborosiloxane) can be synthesized by reacting boric acid with VTEOS or VTMEOS. Comparison of XRD data of ceramics derived from poly(vinylborosiloxane)s synthesized from these two alkoxysilanes will throw some light on the influence of nature of alkoxy substitutent on silicon on the ceramics formed. Direct comparison has been possible only for the heat treatment at 900 °C and 1500 °C as VTMEOS-based system was not heat treated at 1650 °C. XRD patterns of ceramics obtained at 900 °C for both the systems indicate that they behave alike forming SiBOC ceramic. However, XRD patterns of the ceramic obtained at 1500 °C suggest that the onset of β-SiC crystallite formation and separation of graphitic carbon phase is more pronounced with VTMEOS-based system than with VTEOS-based system. These observations are attributed to higher carbon content in VTMEOS based system. Comparison of XRD of ceramics obtained from poly(phenylborosiloxane) [21] and poly(vinylborosiloxane) at at 900 °C, 1500 °C and 1650 °C suggests that both the systems behave alike.
FTIR spectra (Fig. 4) of the ceramics obtained from poly (vinylborosiloxane) at 900 °C, 1500 °C and 1650 °C support the conclusions drawn from XRD patterns. Peaks corresponding to Si–O–Si and B–O linkages are observed in the samples heat treated at 900 and 1500 °C. BSiV-1 heat treated at 900 °C shows absorptions at 1379, 1061, 796 and 455 cm−1 due to νB–O, νSi–O–Si, ρSi–O–Si/νSi-C and δSi–O–Si respectively. Similar peaks were observed for BSiV-2 and BSiV-3 samples heated at 900 °C. Absorption at 1194 cm−1 due to νB–OH present in BSiV-3 is absent in the other two samples. For the samples heat treated up to 1500 °C, peak at 1194 cm−1 corresponding to νB–OH seen in BSiV-2 and BSiV-3 is absent for BSiV-1. The peak at 1379 cm−1 due to νB–O for BSiV-1 becomes more prominent and shifts to higher wave numbers, 1415 and 1439 cm−1 for BSiV-2 and BSiV-3 respectively corresponding to νB–O–B bond of borate network structure [60]. This implies that increase in boric acid concentration in the monomer feed favors the formation of borate network structures in addition to SiBOC ceramics.
FTIR spectra of samples heat treated at 1650 °C exhibit strong absorptions corresponding to SiC at 825 cm−1 (for BSiV-1) which result at the cost of peaks corresponding to Si–O–Si and B–O/B–O–B observed for 1500 °C heat treated samples. A shoulder peak at 880 cm−1 for the 1650 °C heat treated samples is due to Si–O–B linkages, evidencing the presence of a residual glassy phase in the ceramic [53, 61].
The weight loss data of the samples at 1500 °C (42–67 wt.%, Table 1) coupled with FTIR and XRD studies suggest that the weight loss is probably due to the loss of low molecular weight products resulting from further ceramization and loss of B2O3. The inference that the mass loss is also due to B2O3 is supported by the fact that BSiV-3, synthesized using the lowest concentration of VTEOS in the monomer feed, shows the maximum weight loss. The weight loss is much higher (74–79 wt.%) for samples heat treated at 1650 °C implying that significant structural changes have taken place with the increase in temperature. FTIR and XRD studies clearly support the formation of β-SiC at 1650 °C. Complete conversion to β-SiC at 1650 °C is due to the dominance of carbothermal reduction involving nano domains of SiO4 and C leading to gaseous byproducts such as SiO, CO and CO2 and loss of B2O3 which account for the increase in mass loss with the increase in heat treatment temperature. In addition, reaction involving amorphous SiO2 and β-SiC leading to the formation of gaseous products SiO and CO cannot be ruled out [58, 62].
The Raman spectra (Fig. 5) show peaks corresponding to Si–C stretching mode of β-SiC at 777 cm−1 and a peak at 940 cm−1 due to the presence of α and β-SiC [63, 64]. Peaks corresponding to characteristic features of disordered graphitic forms of carbon, the D (disordered) band at around 1351 cm−1 and the G (graphite) band at around 1581 cm−1 (BSiV-1). The higher energy G band is assigned to graphitic single crystals and it is caused by the vibrational mode of graphitic layers. In addition, shoulder peaks at 1615 cm−1 are observed in the samples which might be due to G band splitting. These findings suggest that for SiBOC ceramics, significant ordering of free C phase takes place during annealing at 1500 °C. In general the extent of SiC conversion is less in the case of BSiV-3 and BSiV-2 compared to BSiV-1 at 1500 °C. Peaks corresponding to D and G bands seen around 1343 and 1574 cm−1are less intense in the case of 1650 °C heat treated ones and the same trend is observed for all the systems. Second order D band of free carbon found at 2680 cm−1 is found to be feeble as the carbon has been utilized for the carbothermal reduction for the conversion of SiBOC to SiC.

Raman spectra of ceramics obtained at 1500 °C (bottom) and 1650 °C (top) from: a BSiV-1, b BSiV-2, and c BSiV-3
A broad peak below 600 cm−1 in the Raman spectrum of BSiV-3 (Fig. 5c) annealed at 1500 °C could be attributed to the presence of silica or amorphous SiC. The peaks corresponding to the modes of transverse optical (TO) and longitudinal optical (LO) phonons at ~ 780 cm−1 and ~ 920–950 cm−1 in ceramics annealed at 1500 °C characterize the crystalline structure of β-SiC [63, 64]. Both peaks have significant downshift with respect to the TO (796 cm−1) and LO (970 cm−1) photomodes of bulk β-SiC which could either be ascribed to quantum confinement effects due to nanosized crystallite of SiC or due to the stacking faults. Full width at half maximum (FWHM) of TO and LO modes lie in the range of ~ 30–45 cm−1 and ~ 60–70 cm−1 suggesting that these samples contain defects and stacking faults, which are also responsible for the shifts and variation in the peak positions of the TO and LO modes of SiC. BSiV-1 and 3 annealed at 1650 °C exhibit TO peak at 777 and 779 cm−1 and LO peak at 949 and 941 cm−1 respectively indicating the presence of 4H polytype along with β-SiC. An asymmetric peak at 792 cm−1 with a shoulder at ~ 760 cm−1 (Fig. 5b, 1650 °C) arise from SiC polytypism (presence of 6H and 4H SiC) when Brillouin zone is folded due to defect in β-SiC stacking of carbon and silicon layers. From Raman spectral analysis, it could be drawn out that β-SiC crystallites with numerous stacking faults are formed when BSiV is annealed at 1500 °C and further leads to nucleation of α-polytypes at 1650 °C [65].
Structural evolution of the "free’’ carbon phase in BSiV systems was also examined from Raman spectra. The Raman signals corresponding to free carbon arises from the different hybridization states of carbon atoms as well as due to their arrangement within the network. The G band at ~ 1580 cm−1 is generated by in-plane bond stretching of sp2hybridized carbon pairs. The D band at ~ 1340 cm−1 corresponds to the bond stretching of sp3 hybridized carbon pairs. Presence of overtones of D and G bands at ~ 2700 and 2900 cm−1 in BSiV annealed at 1500 °C indicates that the nature of free carbon is nanocrystalline graphite and this observation is also complimented by the G band position at ~ 1580 cm−1. The intensity ratio ID/IG is used to evaluate the carbon cluster size, La, of the free carbon domains, as proposed by Ferrari and Robertson [66]:
C' (λ) is wavelength dependent constant. The cluster size, La of BSiV-1, 2 and 3 at 1500 °C are calculated to be 1.15, 1.59 and 0.94 nm respectively. The cluster size of 1.59 nm in BSiV-2 indicates that the system contains more of sp3 sites. In other words, 1.5 mol of VTEOS when reacted with 1 mol of boric acid produces carbon clusters with more number of sp3 sites which in turn reacts with SiO2 intermediate leading to the formation of SiC. This observation is also complimented by the formation of crystalline SiC with no evident free carbon at 1650 °C. The peak at 1620 cm−1 in BSiV-3 annealed at 1500 °C is assigned as D band induced by disorder in carbon matrix. The peak at 1675, 1708 and 1681 cm−1 in BSiV systems annealed at 1650 °C along with absence of overtones of D and G band point out that the free carbon is diamond like carbon with considerable fraction of sp3 bonds. In the case of BSiV-1, higher VTEOS concentration favors the formation of nanocrystalline diamond like carbon phases as evidenced by the peak centered around 1150 cm−1. The peak positioned in between D and G band for BSiV-1 and BSiV-2 and BSiV-3 annealed at 1650 °C arises due to the incorporation of heteroatom in carbon matrix which in this case is boron/silicon forming an SiBOC ceramic matrix. The summary of the peak positions of c obtained after heat treatment at 1500 °C and 1650 °C are given in Table 2.
29SiMAS-NMR spectra of BSiV pyrolyzed at 900 °C, 1500 °C and 1650 care shown in Fig. 6. At 900 °C BSiV-1 and BSiV-2 shows three peaks at − 31 ppm, a broad peak centred around − 71 ppm and at − 107 ppm which indicate the presence of SiC2O2, SiCO3 and SiO4 environments respectively [53]. At 1500 °C one resonance centered around − 16.4 ppm is prominent which is attributed to β-SiC with minor component of SiO4 indicated by weak resonance at − 107 ppm.

29Si–MAS NMR spectra of ceramics of a BSiV-1, b BSiV-2, and c BSiV-3
Further pyrolysing at 1650 °C shows three distinct resonances at − 15.8, − 20.3, − 24.5 ppm. Earlier studies [67, 68] suggests that α-SiC has three closely spaced resonances at around − 16 ppm (over lapping with β-SiC resonance), − 20 ppm and -26 ppm. Here, the downfield resonance (− 15.8 ppm) is more prominent compared to the other two resonances suggesting higher amount of β-SiC with minor amount of α-SiC. At 900 °C, BSiV-2 shows peaks at − 71 ppm and − 100 ppm indicating the presence of SiCO3 and SiO4 respectively while BSiV-3 shows only one major peak at − 107 ppm due to SiO4 groups [53]. Thus, in pyrolyzed sample at 900 °C, formation of SiO2C2 and SiO3C units were observed in addition to SiO4 groups with increase in VTEOS concentration. This observation suggests that the rearrangements of Si–C and Si–O bonds which occur by nucleation and crystal growth mechanism usually observed in SiOC/SiBOC systems is more favoured in BSiV-1 synthesized using the monomer feed ratio (boric acid:VTEOS) 1:2. It can be seen that at 900 °C, the spectrum is characteristic of a SiOC glassy network with a distribution of SiCxO4–x environments. The pyrolyzed oligomer is a boron modified SiOC phase where SiCO3, SiC2O2, SiC3O, BC2O and BCO2 units can be present. It is difficult to identify boron containing species in the mixed ceramics as there is not much notable difference in the chemical shift values of the Si units to distinguish between -Si–O–Si- and –Si–O–B– bonds [69, 70]. BSiV-2 and BSiV-3 have the similar pattern of peaks compared to BSiV-1 for both 1500 °C and 1650 °C with varying amounts of respective components. At 1500 °C in all the three systems, the peaks are not well defined and distinct, and in addition exhibit a broad peak starting from − 100 ppm indicating the presence of SiO4 units. This clearly indicates that conversion of poly(vinylborosoiloxane) to SiC is not complete at 1500 °C.
At 1650 °C all the three systems show three distinct resonances which facilitate to calculate the ratio of β-SiC to α-SiC. The ratios of β-SiC to α-SiC are 77:23 for BSiV-1 and 72:28 for both BSiV-2 and BSiV-3. At 1650 °C, the above distinction between alpha and beta forms of SiC is not possible in 13C NMR spectra due to broad line shapes (Fig. 7) compared to 29Si-NMR spectra. But one can observe increase in line width from top to bottom which may be attributed to the change in the particle size. However, not much variation in the resonance lines was observed as compared to the previous studies [70]. Here, peak at 23.7 ppm is attributed to β-SiC which is overlapping with the middle resonance of the three peaks 15.4, 23.7 and 29.7 ppm of α-SiC.

13C-MAS NMR of poly(vinylborosiloxane)s heat treatedat 1650 °C
4 Conclusions
Vinyl-functionalized borosiloxane polymers which give high ceramic yield (73–90% at 900 °C) were synthesized by non-aqueous sol–gel process from boric acid and vinyltriethoxysilane. The effect of variation in monomer feed ratio of the precursors upon the microstructure evolution of veramics at 900 °C, 1500 °C and 1650 °C were investigated in detail using spectroscopic techniques. The IR, XRD and NMR spectroscopic analyses revealed the formation of glassy SiOC/SiBOC phases at 900 °C. With increase in heat treatment temperature to 1500 °C, evolution of crystalline SiC begins followed by complete conversion at 1650 °C. Raman spectral analysis also complements the above observation and indicates the presence of free carbon at and above 1500 °C. Variation of boron concentration in the monomer feed ratio had a profound effect on the ceramic yield and on the crystallite size of β-SiC whereas the ratio of β to α-SiC remained unaffected. Heat treatment studies of poly(vinylborosiloxane) at 900, 1500 and 1650 °C suggests that poly(vinylborosiloxane) can serve as an effective precursor for SiBOC ceramics up to 1500 °C. At 1650 °C, SiC is obtained in poor yield (21–26%) and hence, would result in highly porous ceramic structure if poly(vinylborosiloxane) is used as a precursor for SiC matrix. By carefully fine tuning the processing step of the non-aqueous sol–gel process, soluble poly(vinylborosiloxane) useful for application as ceramic adhesives, ceramic coatings and matrix resins for ceramic matrix composites can be obtained.
References
J. Bill, F. Aldinger, Precursor-Derived Covalent Ceramics, in Precursor-Derived Deramics, ed. by J. Bill, F. Wakai, F. Aldinger (Wiley-VCH, Weinheim, Federal Republic of Germany, 1999), pp. 32–51
E. Ionescu, H.J. Kleebe, R. Riedel, Silicon-containing polymer-derived ceramic nanocomposites (PDC-NCs): preparative approaches and properties. Chem. Soc. Rev. 41, 5032 (2012)
P. Colombo, G. Mera, R. Riedel, G.D. Soraru, Polymer-derived ceramics: 40 years of research and innovation in advanced ceramics. J Am. Ceram. Soc. 93, 1805–1837 (2010)
P. Colombo, R. Raj, M. Singh, Advances in Polymer Derived Ceramics and Composites: Ceramic Transactions (John Wiley & Sons, New Jersey, 2010)
P. Colombo, R. Riedel, G.D. Soraru, H.J. Kleebe, Polymer Derived Ceramics: From Nano-Structure to Applications (Destech Publications Inc, Lancaster, 2010)
I. Emanuel, G. Mera, R. Riedel, Polymer-Derived Ceramics (PDCs): Materials Design Towards Applications at Ultrahigh-Temperatures and in Extreme Environments: In Nanotechnol. Concepts, Methodol. Tools, Appl. IGI Global, 2014, pp. 1108–1139.
G. Mera, M. Gallei, S. Bernard, E. Ionescu, Ceramic nanocomposites from tailor-made preceramic polymers. Nanomaterials 5, 468–540 (2015)
J.O.B. Rivera, M.H. Talou, Y.M.X.H. Hung, M.A. Camerucci, Study of a silicon-based preceramic for the processing of polymer-derived ceramics. J. Sol-Gel Sci. Tech. 91, 446–460 (2019)
C. Stabler, E. Ionescu, M. Graczyk-Zajac, I. Gonzalo-Juan, R. Riedel, Silicon oxycarbide glasses and glass-ceramics: “All-Rounder” materials for advanced structural and functional applications. J. Am. Ceram. Soc. 101, 4817–4856 (2018)
Y. Blum, G.D. Sorarù, A.P. Ramaswamy, D. Hui, S.M. Carturan, Controlled mesoporosity in SiOC via chemically bonded polymeric “Spacers”. J. Am. Ceram. Soc. 96, 2785–2792 (2013)
A.D. Chomel, P. Dempsey, J. Latournerie, D. Hourlier-Bahloul, U.A. Jayasooriya, Gel to glass transformation of methyltriethoxysilane: a silicon oxycarbide glass precursor investigated using vibrational spectroscopy. Chem. Mater. 17, 4468–4473 (2005)
B.V.M. Kumar, Y.W. Kim, Processing of polysiloxane-derived porous ceramics: a review. Sci. Technol. Adv. Mater. 11, 1–16 (2010)
D. Erb, K. Lu, Effect of additive structure and size on SiO2 formation in polymer-derived SiOC ceramics. J. Am. Ceram. Soc. 101, 5378–5388 (2018)
J. Ma, L. Shi, Y. Shi, S. Luo, J. Xu, Pyrolysis of polymethylsilsesquioxane. J. Appl. Polym. Sci. 85, 1077–1086 (2002)
M. Sitarz, C. Czosnek, P. Jeleń, M. Odziomek, Z. Olejniczak, M. Kozanecki, J.F. Janik, SiOC glasses produced from silsesquioxanes by the aerosol-assisted vapor synthesis method, Spectrochim. Acta Part A: Mol. Biomol. Spectrosc. 112, 440–445 (2013)
P.R. Aravind, G.D. Soraru, Porous silicon oxycarbide glasses from hybrid ambigels. Micro. Meso. Mat. 142, 511–517 (2011)
M.A. Schiavon, C. Gervais, F. Babonneau, G.D. Soraru, Crystallization behavior of novel silicon boron oxycarbide glasses. J. Am. Ceram. Soc. 87, 203–208 (2004)
D. Devapal, S. Packirisamy, P. V. Prabhakaran, K. J. Sreejith, A. Paul, A. Painuly, Process for solventless synthesis of resinous borosiloxane oligomer precursors for ceramics, Indian Patent 277874 (2016).
T.S. Sasikala, D. Thomas, D. Devapal, Studies on evolution of nano SiC ceramics from allylborosiloxane. Ceram. Int. 41, 1618–1626 (2016)
T.S. Sasikala, D. Devapal, Studies on high temperature evolution of polymer derived nano SiC ceramics. Mater. Sci. Forum 830, 493–497 (2015)
D. Devapal, S. Packirisamy, K.J. Sreejith, P.V. Ravindran, B.K. George, Synthesis, characterization and ceramic conversion studies of borosiloxane oligomers from phenyltrialkoxysilanes. J. Inorg. Organomet. Polym. 20, 666–674 (2010)
K.J. Sreejith, P.V. Prabhakaran, K.P. Laly, R. Dimple, S. Packirisamy, Vinyl-functionalized poly(borosiloxane) as precursor for SiC/SiBOC nanocomposite. Ceram. Int. 42, 15285–15293 (2016)
S. Rubinsztajn, New facile process for synthesis of borosiloxane resins. J. Inorg. Organomet. Polym. Mater. 24, 1092–1095 (2014)
V. Vijay, S. Bhuvaneswari, V.M. Biju, R. Devasia, Influence of titanium silicide active filler on the microstructure evolution of borosiloxane-derived Si–B–O–C ceramics. J. Ceram. Sci. Tech. 07, 97–106 (2016)
V. Vijay, V.M. Biju, R. Devasia, Active filler controlled polymer pyrolysis—a promising route for the fabrication of advanced ceramics. Ceram. Int. 42, 15592–15596 (2016)
A. Klonczynski, G. Schneider, R. Riedel, R. Theissmann, Influence of boron on the microstructure of polymer derived SiCO ceramics. Adv. Eng. Mater. 6, 64–68 (2004)
A.H. Tavakoli, R. Campostrini, C. Gervais, F. Babonneau, J. Bill, G.D. Sorarù, A. Navrotsky, Energetics and structure of polymer-derived Si–(B–)O–C glasses: effect of the boron content and pyrolysis temperature. J. Am. Ceram. Soc. 77, 303–309 (2014)
S. Hoshii, A. Kojima, S. Otani, Mechanical properties and oxidation resistivity of carbon fiber/ceramic composites prepared from borosiloxane. J. Mat. Res. 11, 2536–2540 (1996)
R.L. Siqueira, I.V.P. Yoshida, L.C. Pardini, M.A. Schiavon, Poly(borosiloxanes) as precursors for carbon fiber ceramic matrix composites. Mat. Res. 10, 147–151 (2007)
K. J. Sreejith, S. Packirisamy, Phenylborosiloxane-Derived Ceramic Matrix Composites, High Temperature Ceramic Materials and Composites, Eds. W. Krenkel, J. Lamon. Aviso VerlagsgesellschaftmbH, Berlin, 2010, pp. 712–718.
B. Swaminathan, A. Painuly, S. K. Manwatkar, S. Packirisamy, Preceramic polymer derived C/CSiC and C/C-SiBCO composites for high temperature applications in High Temperature Ceramic Materials and Composites, Eds. W. Krenkel and J. Lamon. AvisoVerlagsgesellschaftmbH, Berlin, 2010, pp. 724–730.
K.J. Sreejith, A. Painuly, B.V. Rajasekhar, P.P. Shyin, V. Vijay, R. Devasia, P.V. Prabhakaran, S. Packirisamy, A process for polymer-derived Cf/SiBOC ceramic matrix composites, Indian Patent Appl. 201841020417 (2018).
V. Vijay, S. Siva, K.J. Sreejith, P.V. Prabhakaran, R. Devasia, Effect of boron inclusion in SiOC polymer derived matrix on the mechanical and oxidation resistance properties of fiber reinforced composites. Mat. Chem. Phys. 205, 269–277 (2018)
S.G. Nair, K.J. Sreejith, S. Packirisamy, T.G. Babu, R. Devasia, Polymer derived PyC interphase coating for C/SiBOC composites. Mat. Chem. Phys. 204, 179–186 (2018)
R. Devasia, S. G. Nair, K. J. Sreejith, S. Packirisamy, Fibre-reinforced ceramic matrix composite material with polymer derived interphase coating. Indian Patent No. 299956 (2018).
G. T. Burns, G. A. Zank, High density silicon carbide sintered bodies from borosiloxanes, US Patent 5,112,779 (1992).
D. Devapal, M. P. Gopakumar, P. V. Prabhakaran, S. Packirisamy, A process for preparation of silicon carbide coated carbon nano-materials using polyborosiloxanes, Indian Patent Appl. 2017–41024214 (2017)
R.P. Alonso, G.D. Soraru, Synthesis and characterization of hybrid borosiloxane gels as precursors for Si–B–O–C fibers. J. Sol-Gel Sci. Tech. 43, 313–319 (2007)
H.W. Bai, G. Wen, X.X. Huang, Z.X. Han, B. Zhong, Z.X. Hu, X.D. Zhang, Synthesis and structural characterization of SiBOC ceramic fibers derived from single-source polyborosiloxane. J. Eur. Ceram. Soc. 31, 931–940 (2011)
A. Tamayo, R.P. Alonso, F. Rubio, J. Rubio, J.L. Oteo, Synthesis and characterization of boron silicon oxycarbide glass fibers. J Non-Cryst Solids 358, 155–162 (2012)
K.J. Sreejith, T. Fey, P. Greil, Siliconboronoxycarbide (SiBOC) foam from methyl borosiloxane. Ceram. Trans. 243, 47–60 (2014)
N. Tohge, A. Matsuda, T. Minami, Coating films of 20B2030.80Si02 by the sol-gel method, J. Am. Ceram. Soc., 70 (1987) C13–C15.
M.A. Villegas, J.M.F. Navarro, Characterization of B2O3–SiO2 glasses prepared via sol-gel. J. Mat. Sci. 23, 2464–2478 (1988)
Y. Abe, T. Gunji, Y. Kimata, M. Kuramata, A. Kasgoz, T. Misono, Preparation of polymetalloxanes as a precursor for oxide ceramics. J. Non-Cryst. Solids 121, 21–25 (1990)
A. Kasgoz, T. Misono, Y. Abe, Preparation and properties of polyborosiloxanes as precursors for borosilicate formation of Si02–B203 gel fibers and oxides by the sol–gel method using tetraacetoxysilane and borontri-n-butoxide, J. Polym. Sci. A, Polym. Chem. 32 (1994) 1049–1056.
G. Ambadas, S. Packirisamy, K.N. Ninan, Synthesis, characterization and thermal properties of boron and silicon containing preceramic oligomers. J. Mat. Sci. Lett. 21, 1003–1005 (2002)
S. Packirisamy, G. Ambadas, P.K. Narendranath, K. N. Ninan, A process for the synthesis of boron and silicon containing preceramic oligomers. Indian Patent No. 208583(2007).
D. Devapal, Studies on inorganic and organometallic polymers (Ph.D. Thesis), Mahatma Gandhi University, India, 2007.
P. V. Prabhakaran, Studies on non-oxide ceramics derived from polymers and their applications (Ph.D. Thesis), University of Kerala, India, 2008.
K. J. Sreejith, Polymer derived ceramics and their high temperature applications (Ph.D. Thesis), University of Kerala, India, 2010.
G. D. Soraru, F. Babonneau, C. Gervais, N. Dallabona, Hybrid RSiO1.5/B2O3 gels from modified silicon alkoxides and boric acid, J. Sol–Gel Sci. Technol. 18 (2000) 11–19.
F. D. Snell, C. L. Hilton (eds.), in Encyclopedia of Industrial Chemical Analysis, vol. 7 (Interscience Publishers, New York, 1968, pp. 324.
C. Gervais, F. Babonneau, N. Dallabonna, G.D. Soraru, Sol–gel-derived silicon-boron oxycarbide glasses containing mixed silicon oxycarbide (SiCxO4_x) and Boron Oxycarbide (BCyO3_y) Units. J. Am. Ceram. Soc. 84, 2160–2164 (2001)
R.A. Mantz, R.F. Jones, K.P. Chaffee, J.D. Lichtenhan, J.W. Gilman, M.K. Ismail, M.J. Burmeister, Thermolysis of polyhedraloligomeric silsesquioxane (POSS) macromers and POSS−siloxanecopolymers. Chem. Mater. 8, 1250–1259 (1996)
G.D. Soraru, F. Babonneau, S. Maurina, J. Vicens, Sol-gel synthesis of SiBOC glasses. J. Non Cryst. Solids 224, 173–183 (1998)
A.M. Wootton, M. Rappensberger, M.H. Lewis, S. Kitchin, A.P. Howes, R. Dupree, Structural properties of multicomponent SiOC glasses derived from metal alkoxide precursors. J Non-Cryst Solids 204, 217–227 (1996)
R. Dhiman, V. Petrunin, K. Rana, P. Morgen, Conversion of wooden structures into porous SiC with shape memory synthesis. Ceram. Int. 37, 3281–3289 (2011)
A. Saha, R. Raj, Crystallization maps for SiCO amorphous ceramics. J. Am. Ceram. Soc. 90, 578–583 (2007)
P. Scherrer, Bestimmung der Grosse und der inneren Struktur von Kolloidteilchen mittels Rontgenstrahlen. Nachr. Ges. Wiss. Gottingen 26, 98–100 (1918)
M.A. Schiavon, N.A. Armelin, I. Valéria, P. Yoshida, Novel poly(borosiloxane) precursors to amorphous SiBCO ceramics. Mater. Chem. Phy. 112, 1047–1054 (2008)
G.D. Soraru, N. Dallabona, C. Gervais, F. Babonneau, Organically modified SiO2–B2O3 gels displaying a high content of borosiloxanes (B−O−Si) bonds. Chem Mater. 11, 910–919 (1999)
D.H. Filsinger, D.B. Bourrie, Silica to silicon: key carbothermic reactions and kinetics. J. Am. Ceram. Soc. 73, 1726–1732 (1990)
G. Gouadec, P. Colomban, Non-destructive mechanical characterization of SiC fibers by Raman spectroscopy. J. Eur. Cer. Soc. 21, 1249–1259 (2001)
R. Dhiman, E. Johnson, E.M. Skou, P. Morgen, S.M. Andersen, SiC nanocrystals as Pt catalyst supports for fuel cell applications. J. Mater. Chem. A 1, 6030–6036 (2013)
H.P. Martin, E. Muller, G. Irmer, F. Babonneau, Crystallization behaviour and polytype transformation of polymer derived Silicon carbide. J. Eur. Cer. Soc. 17, 659–666 (1997)
A.C. Ferrari, J. Robertson, Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 61(20), 14095–14107 (2000)
G.W. Wagner, B.K. Na, M.A. Vannice, High resolution solid state NMR of 29Si and 13C in β-Silicon carbides. J. Phys. Chem. 93, 5061–5064 (1989)
X. Xie, Z.Yang, R. Ren, Leon L. Shaw, Solid state 29Si magic angle spinning NMR: investigation of bond formation and crystallinity of silicon and graphite powder mixtures during high energy milling, Mater. Sci. Eng. A255 (1998) 39–48.
A.D. Irwin, J.S. Holmgren, T.W. Zerda, J. Jonas, Spectroscopic investigation of Borosiloxane bond formation in the sol-gel process. J. Non-cryst. Solids 89, 191–205 (1987)
A.D. Irwin, J.S. Holmgren, J. Jonas, Solid state 29Si and 11B NMR studies of sol gel derived borosilicates. J. Non-cryst. Solids 101, 249–254 (1988)
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Devapal, D., Sreejith, K.J., Swaminathan, B. et al. Influence of Heat Treatment Temperature on the Microstructure Evolution of Poly(vinylborosiloxane) Derived Ceramics. J Inorg Organomet Polym 30, 2224–2233 (2020). https://doi.org/10.1007/s10904-020-01457-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-020-01457-1