
Abstract
This study is aimed at the present challenge in self-controlled catalysts, addressing how to furnish the catalysts with cascade/non-cascade-switchable catalytic ability. By borrowing the “soft” properties and autonomous ability from nature, this objective was met by reporting an artificial polymer reactor that was composite of tri-layer architectures made of two inversely-thermosensitive outward layers and a non-responsive middle layer. With the middle layer containing catalytic metal nanoparticles, the two thermosensitive outward layers consisted separately of a negatively-temperature responsive polymer and a positively-temperature responsive composite. The opposite responsiveness at the two thermosensitive outward layers induced convex/flat/concave-switchable shapes in this reactor, leading to cascade/non-cascade-alterable substrate channeling to the reactive middle layer. In this way, this polymer reactor led to the occurrence of cascade/non-cascade-switchable catalytic ability. This design shares a promising prospect with the struggling field of self-controlled catalysts, which allows opportunities to finely control catalytic processes.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The development of self-controlled catalysts remains an attractive subject in chemical synthesis. The use of self-controlled catalysts in chemical synthesis would allow the ongoing reactions to run in a pre-planned way, decreasing the synthesis and separation steps and accordingly making possible one-pot synthetic-ability [1, 2]. This privilege often arises from the careful modulation of the structure–activity relationship at the undergoing catalysts, which leads to either controlled access for substrate or micro-phase separation from the catalytic systems. In this way, the use of self-controlled catalysts leads to the occurrence of self-controlled catalytic behaviors. Despite the progress [3, 4], the applications of self-controlled catalysts in practical catalytic processes have been not remarkable over the years. One important hindrance lies in the diversity of the practical catalytic processes and the complication of the catalytic mechanisms, which often incorporate both cascade and non-cascade processes [5, 6]. It is not realistic to achieve such self-controlled catalysts capable of concurrently meeting both cascade and non-cascade processes by basing on currently available methods and technology. As such, new methods and technology are urged.
Nature has been mankind’s mother for knowledge. By the perfect functionality and autonomous ability, the nature has been providing inspirations to resolve complicated issues. Among these are biological systems, which share a prospect with the struggling field of self-controlled catalysts. Natural biological systems (mainly active species and entities), as catalytic reactors, often have the abilities to fit for multi-pathway reaction processes (typically catalytic hydrolysis and redox reactions) [7, 8], given that these abilities are essential for their survival. The clue behind this can be related to the constituent components in these natural reactors, which are often polymeric and “soft” in nature and tolerate an “arbitrary” piling-up of the morphology and structures. The “soft” properties and “arbitrary” piling-up of the morphology and structures allow a fine modulation of the substrate channeling to the internal active species, which thereby makes possible the switching from simple reactions to cascade processes. In this way, natural biological systems, as catalytic reactors, show the fine self-controlled ability to meet complicated reaction conditions and key challenges.
Inspired by the principle in nature, we herein address the present challenge by reporting an artificial polymer reactor capable of cascade/non-cascade-switchable catalytic ability. By borrowing the “soft properties” and autonomous ability from nature, this polymer reactor was fabricated with tri-layer architectures that allowed finely controlling the substrate channeling to the active middle layer. As outlined in Scheme 1, the first layer consisted of poly(N-isopropylacrylamide) (PNIPA), which was capable of negatively-temperature responsive properties (i.e., the increasing temperature leads to shrinking polymeric networks) and accordingly allowed open/closed access to the coupled second layer (i.e., the middle layer). The second layer was composed of poly(acrylamide) (PAAm) and encapsulated metal nanoparticles, capable of catalytic reduction. In contrast, the third layer was a positively-temperature responsive polymer composite of poly(2-acrylamido-2-methylpropanesulfonic acid) (PAMPS) and a stoichiometric ratio of poly(1-vinylimidazole) (PVI) that exhibited switchable interactions [9, 10]. The self-healing and dissociation between PVI and PAMPS induced switchable networks in the third layer, opening or closing by depending on the temperature. Such switchable networks, in combination with the catalytic properties of acidic PAMPS, enabled the third layer to proceed with hydrolytic activities except validating access (typically for ester compounds and their hydrolysis). In integration with these individual properties, the tri-layer architectures in this reactor would cooperatively work for the occurrence of the cascade/non-cascade-switchable process. At relatively low temperatures (lower than the transition temperature of PNIPA; cf. Pathway 1), both the first-layer and second-layer (but not the third layer due to the closed polymeric networks) would work, which allowed the engaging substrate to run with simple reactions (such as catalytic reduction). In contrast, at relatively high temperatures (higher than the transition temperature of PVI-PAMPS; cf. Pathway 2), both the third-layer and second-layer (but not the first layer due to the closed networks) would work, which allowed the substrate to run with cascade reactions (typically, cascade hydrolysis and reduction). There was almost no reaction available in between, given the closed networks in both the thermosensitive outward layers. In this way, this polymer reactor demonstrated the unique cascade/non-cascade-switchable catalytic ability.

Proposed mechanism for the cascade/non-cascade-switchable catalytic ability at the artificial reactor (in which SVA represents the PVI-PAMPS layer, AmAg means poly(acrylamide) containing Ag nanoparticles, and SI stands for the PNIPA layer) (Color figure online)
To that end, bis(2,4-dinitrophenyl)oxalate (DNPO) was elaborately selected as the testing substrate, given that the compound can be subject to not only a simple reaction (such as catalytic reduction) but also a catalytic cascade process (such as cascade hydrolysis and reduction) [11], in which the former would result in the formation of bis(2,4-diaminophenyl)oxalate (DAPO) and the latter would lead to 2,4-diaminophenol (DAP). The objective of this study is to demonstrate that functional catalysts capable of cascade/non-cascade-switchable catalytic ability can be realized by developing artificial polymer reactors, which accordingly suggests opportunities to finely control catalytic processes.
2 Experimental Section
2.1 Preparation of Reactors
The chemicals used are of analytic grade and were used as received from Sigma-Aldrich. The artificial reactor, as explained, was prepared with polymeric tri-layer architectures that were composite of two switchable outward layers and a non-responsive middle layer. To connect these layers [12, 13], the middle layer was first prepared with polymerization and then exposed separately by two sides to two different molecular solutions intended for preparing the switchable outward layers, which allowed polymerizable monomers in the solutions to diffuse into the middle layer. The molecular solutions were then polymerized with the addition of a radical initiator to form polymeric networks interpenetrated with the middle layer. In this way, these layers were connected. In detail, acrylamide (1.42 g), silver nitrate (0.28 g), ammonium persulfate (54 mg) and N,N′-methylenebisacrylamide (0.1 g) were dissolved in dimethylsulfoxide (10 ml). After being dispersed and deoxygenated with sonication and nitrogen, the mixture system was kept overnight at 60 °C for complete polymerization to form the middle layer. The encapsulated Ag ions were subsequently reduced with an excess of sodium borohydride. Following this, one side of the middle layer was exposed to the molecular solution intended for preparing the PNIPA layer, in which N-isopropylacrylamide (2.26 g), ammonium persulfate (54 mg) and N,N′-methylenebisacrylamide (0.1 g) were dissolved in dimethylsulfoxide (10 ml). Polymerization was performed at 60 °C for 2 h to fix the layer. After this, the other side of the middle layer was exposed to the molecular solution intended for preparing the PVI-PAMPS layer, where 2-acrylamido-2-methylpropanesulfonic acid (AMPS; 2.655 g) and a certain amount of 1-vinylimidazole (VI) (discussed afterward), along with ammonium persulfate (81 mg) and N,N′-methylenebisacrylamide (0.1 g), were dissolved in dimethylsulfoxide (10 ml). Polymerization was then performed again at 60 °C for 2 h to fix the layer. In this way, the artificial tri-layer reactor was prepared (named “SVA-AmAg-SI”; herein, “S” means switchable properties, “V” shows PVI, “A” stands for PAMPS, “Am” shows PAAm, “Ag” means Ag nanoparticles, and “I” represents PNIPA).
Given the dominance of the thermosensitive outward layers in the self-controlled ability of the artificial reactor, three control groups which were lack of either one or two thermosensitive outward layers were also prepared under comparable conditions (i.e., SVA-AmAg-NT, NT-AmAg-SI and NT-AmAg-NT). By replacing the thermosensitive layers with non-responsive polystyrene (marked by “NT”), these control groups therefore allowed the rest of layers to work and accordingly provided a straightforward profile for Pathways 1 and 2 (cf. Scheme 1). In this way, these control groups, along with the artificial reactor, were prepared.
2.2 Characterization
The FTIR spectra of the prepared reactors were recorded using a Nicolet MX-1E apparatus (USA). The TEM images were obtained using a JEM-2100 transmission electron microscope (TEM) (Japan). The absorption bands of the surface plasma resonance (SPR) were determined using a UV-2700 spectrophotometer (Japan). The energy-dispersive spectra (EDS) were obtained using a MIRA3-XMU apparatus (USA). The change of the shape upon temperature was recorded with a digital camera in which the slices of the prepared reactors were immersed in water.
2.3 Thermosensitive Transition
The thermosensitive transition at the switchable outward layers of the prepared reactors was studied as a function of temperature by using dynamic light scattering (DLS) (Zetasizer, UK). For equilibrium, the samples scraped from the thermosensitive switchable layers were kept at specified temperatures in water for at least 5 min before scanning the dynamic radius (Rd). By a comparison between the switchable layers and corresponding non-responsive layers, the change of the dynamic radius reflected the contribution of the thermosensitive transition [14].
2.4 Catalytic Properties
The catalytic properties of the prepared reactors were evaluated in batch formats [15]. The initial concentration of DNPO was 0.025 μmol ml−1 (20 ml PBS; pH 7.0) (NaBH4, tenfold in contrast to DNPO). The content of the reactors used in each testing was 0.1 mg ml−1. The catalytic behavior was spectrophotometrically monitored using a UV-2700 spectrophotometer (Japan) and the catalytic activities were determined from the average of triple runs. Given the potential effect of spontaneous reactions on the catalytic process, the reactions of DNPO without having any reactors were also performed under the comparable conditions and accordingly the effect was deducted from the reported overall activities of these reactors.
2.5 Desorption Electrochemistry
Desorption electrochemistry was further performed to acquire information on the catalytic mechanisms in the prepared reactors [16, 17]. Using an electrochemical workstation equipped with a conventional three-electrode configuration (Au-plate working electrode, Pt-wire counter electrode and Ag/AgCl reference electrode), the prepared reactors (10 mg) which pre-adsorbed with about 2 mmol of substrate were placed in the electrochemical cells encircled by a diffusion-eliminating sonication apparatus (supporting electrolyte: 10 ml PBS; pH 7.0). The desorbing behavior of the absorbed substrate was monitored by circularly scanning the system until a stable desorption profile was reached (scanning range, + 0.3 ~ − 0.9 V; scanning rate, 1 mV s−1).
3 Results and Discussion
3.1 PVI-PAMPS Interactions and Optimization
As explained, the artificial reactor SVA-AmAg-SI functioned by relying on the cooperation between the two thermosensitive outward layers, in which the complementary interactions between PVI and PAMPS in the third layer acted as a molecular switch for the occurrence of catalytic cascade ability. For the switching, it has been known that an excess in either of the constituent VI and AMPS would result in a steric mismatch which causes unsaturated interactions and defective responsiveness (and even no responsiveness, at all) [18], and that only the stoichiometric interactions can lead to the complete association/dissociation and accordingly the best switchable responsiveness. Considering the interactions that can induce an alterable electronic-transition at valent electrons and accordingly a change in UV titration, as shown in often cases [9, 10], the complementary interactions between PVI and PAMPS herein were also studied with UV titration. As shown in Fig. 1, the titration of VI to AMPS resulted in a shift in the UV spectrum. The shift gained a maximal value when the titrated VI reached a critical amount (corresponding to 1.28 mol mol−1 AMPS/VI ratio). Beyond the critical amount, there was no more shift available in the UV spectrum (except increasing absorbance). This outcome indicates that the complementary interactions were saturated by the stoichiometric titration and accordingly the AMPS/VI ratio was optimized. As such, 0.94 g (10 mmol) of VI was used to prepare the artificial reactor SVA-AmAg-SI and its control groups, where 2.655 g (12.8 mmol) of AMPS was adopted.

UV spectra shifting as a function of VI-TFMA ratios by using UV titration (in which, VI (77.28 μmol ml−1; 76 μl) was titrated into AMPS (2.5 μmol ml−1; 3 ml) (Color figure online)
3.2 FTIR, EDS, TEM and SPR Analyses
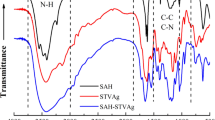
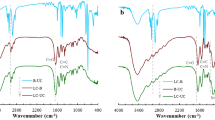
In conjunction with the study of Sect. 3.1, the artificial reactor SVA-AmAg-SI was prepared with polymeric tri-layer architectures that were composite of two thermosensitive outward layers and a non-responsive middle layer. With the middle layer made of PAAm containing metal nanoparticles (i.e., AmAg layer), the two thermosensitive layers consisted separately of PNIPA and PVI/PAMPS (i.e., SI layer and SVA layer). FTIR spectra are first used to track the polymeric composition in these prepared layers. As shown in Fig. 2, three major bands (2950–3700, 1600–1800, and 900–1350 cm−1) appeared in the spectra of these layers, plus an extra band (1400–1600 cm−1) in the SVA layer. These absorption bands were composite because of the multi-component composition, spectroscopically corresponding to the stretching vibration of O–H (N–H), C=O, and C–N (C–C) bonds in these layers [19], plus an extra S=O bond in the SVA layer [20]. The exhibition of these absorption bands from these layers may be ascribed to the constituent components (i.e., PNIPA in the first layer, PAAm in the second layer, and PVI/PAMPS in the third layer) which all include O–H (N–H), C=O, and C–N (C–C) bonds. The presence of S=O bands, along with these major bands in the third layer, revealed the synthetic composition in the third layer, composite of PVI and PAMPS. For further addressing this, we also include the FTIR spectra of these control groups in Fig. 2. By replacing the thermosensitive outward layers with polystyrene, the absorption bands responsible for C=O and S=O bonds disappeared from the outward layers in these control groups. These outcomes indicate that the artificial reactor was the polymer reactor composite of the desired polymeric composition.

FTIR spectra of the three functional layers in the prepared reactors (a SI layer; b AmAg layer; c SVA layer) (Color figure online)
The EDS analysis supported the characterization of the FTIR spectra and agreed with the constituent components in these reactors, which separately exhibited the categorical elements of Ag, C, N, O and S (Fig. 3). Ag nanoparticles with a size of ~ 18 nm were encapsulated in these reactors (Fig. 4) and accordingly the Ag loadings achieved in these reactors were 2.2 wt%. The presence of Ag nanoparticles in these reactors was further supported from the SPR absorption bands which exhibited the typical SPR peaks of Ag nanoparticles at ~ 420 nm (Fig. 5) [21]. Figure 6 displays the digital picture of SVA-AmAg-SI. This artificial reactor demonstrated the expected tri-layer structures. Hence, these reactors were prepared in the desired form.

EDS spectra of the prepared reactors (a SI layer; b AmAg layer; c SVA layer) (Color figure online)

TEM images of metal nanoparticles encapsulated in the prepared reactors (a SVA-AmAg-SI; b NT-AmAg-NT; c SVA-AmAg-NT; d NT-AmAg-SI)

SPR spectra of the prepared reactors (Color figure online)

Digital image of the outward appearance of a slice of SVA-AmAg-SI
3.3 Change of Shape and Thermosensitive Properties
Figure 7 displays the change of the artificial reactor upon changing temperature. With increasing temperature, the slice of this reactor appeared to be convex at relatively low temperatures (such as 25 °C), following by flat at a modest temperature (such as 35 °C) and then concave at higher temperatures (such as 45 °C). The change in the shape was reversible and this reactor can be made to switch back-and-forth among convex, flat and concave shapes upon changing temperature. The thermosensitive properties in the switchable layers led to alterable shapes in the resulting reactor. This outcome, as explained, can be due to the opposite responsiveness in the two thermosensitive outward layers. The opposite responsiveness in the two outward layers may induce asymmetric swelling and shrinkage in this reactor and as a result this reactor demonstrated alterable shapes.

Shape changing with temperature at the artificial reactor SVA-AmAg-SI (a 25 °C; b 35 °C; c 45 °C)
To further address the thermosensitive properties, the dynamic radius of the samples scraped from the thermosensitive outward layers was studied as a function of temperature. As shown in Fig. 8a and b, the dynamic radius (Rd) of both the thermosensitive layers showed significant dependence on temperature, in contrast to the non-responsive PAAm and NT layers which did not have actual dependence on temperature. The dynamic radius of the SI layer decreased with increasing temperature and the major change appeared at ca. 32 °C. Below 32 °C, the SI layer showed relatively a large radius nearly comparable to PAAm that ran with open polymeric networks; above 32 °C, the dynamic radius of the SI layer dramatically decreased, however, to a level almost comparable to that of the NT layer which ran with closed networks. The SI layer evidently showed the negatively-temperature responsive properties. In contrast, the thermosensitive properties of the SVA layer was remarkably different from that of the SI layer, in which the dynamic radius increased with increasing temperature and the major change appeared at ca. 39 °C. Below 39 °C, the SVA layer showed relatively a small radius associating with the complementary interactions between PVI and PAMPS, which intended to shut down the polymeric networks; above 39 °C, the dynamic radius of the SVA layer dramatically increased in response to the dissociation of the complementary interactions, which led to open networks. The SVA layer exhibited the positively-temperature responsive properties, instead. In the zone between two major changes (such as 35 °C), the change in the polymeric networks was actually small and accordingly this reactor did not show an essential change in the shape. These outcomes demonstrate that the alterable shape in the resulting reactor was a result of the cooperation between both the thermosensitive layers. In integration with the oppositely-switchable properties of the two thermosensitive layers which either admitted or denied access to the reactive AmAg layer, one can expect that this reactor would admit alterable substrate channeling and as a result the occurrence of cascade/non-cascade-switchable catalytic ability.

DLS spectra with dynamic-radius changing as a function of temperature (a Differential; b Normal) (Color figure online)
3.4 Cascade/Non-cascade-Switchable Catalytic Ability
The catalytic properties of these reactors are presented in Fig. 9. Three representative temperatures, i.e., 25, 35 and 45 °C, either higher or lower than the transition temperatures of SVA-AmAg-SI (i.e., 32 and 39 °C) (cf. Fig. 8), were selected to scrutinize the switchable catalytic behaviors. The selection of such temperatures was to ensure the switchable zones that can be covered (cf. Scheme 1). Despite the temperature, NT-AmAg-NT showed the lowest activities among these reactors due to the two outward NT layers which largely inhibited access to the reactive middle layer. In contrast, the catalytic activities in other reactors and particularly in the artificial reactor SVA-AmAg-SI showed essential dependence on temperature. At 25 °C, SVA-AmAg-SI showed catalytic activities almost comparable to that in NT-AmAg-SI which functioned with only the first layer and the second layer (i.e., only the SI layer and the middle layer worked). At 45 °C, the catalytic activities in SVA-AmAg-SI became, however, almost comparable to that in SVA-AmAg-NT which functioned with only the third layer and the second layer (i.e., only the SVA layer and the middle layer worked). The artificial reactor SVA-AmAg-SI showed the desired switchable behaviors.

Catalytic activities of the prepared reactors (a 25 °C; b 35 °C; c 45 °C) (in which the similar catalytic activities between SVA-AmAg-SI and controls were marked by a circlet) (Color figure online)
To further address the catalytic switchable behaviors, Fig. 10 displays the UV spectroscopic analysis on the catalytic process of SVA-AmAg-SI. At 25 °C (Fig. 10a), the catalytic process of SVA-AmAg-SI behaved like that of NT-AmAg-SI which led to a decreasing peak for DNPO (360 nm) and meantime an increasing peak for DAPO (302 nm), exhibiting the straightforward reduction from DNPO to DAPO. At 45 °C (Fig. 10c), the catalytic process of SVA-AmAg-SI behaved, however, like that of SVA-AmAg-NT which led to a newly-increasing peak for DAP (290 nm), exhibiting the cascade hydrolysis and reduction of DNPO. The change in the spectra was repeatable after re-switching the temperature between 25 and 45 °C. There was no significant catalytic process available at 35 °C in this artificial reactor (Fig. 10b), given the closed networks in both the thermosensitive outward layers. Once again, this outcome indicates that the desired cascade/non-cascade-switchable catalytic ability occurred in the artificial reactor.

Changing UV/vis spectra with the reduction and/or hydrolysis of DNPO in the prepared reactors (a SVA-AmAg-SI/NT-AmAg-SI at 25 °C; b SVA-AmAg-SI at 35 °C; c SVA-AmAg-SI/SVA-AmAg-NT at 45 °C) (Color figure online)
3.5 Dynamic Binding and Alterable Substrate Channeling
As far as polymer reactors and alterable substrate channeling are concerned, the fundamental issues are known to lie in the interactions between the switchable moieties and substrate [16, 17]. Hence, addressing the interactions between the switchable layers and substrate is often a key for unfolding alterable substrate channeling and accordingly the catalytic mechanisms in polymer reactors. As such, the desorption electrochemistry needs to be performed to gain information in the switchable layers of the artificial reactor. For this point, it has been known that the potential to reduce/oxidize a binding molecule is essentially dependent on the binding strength. Relatively, stronger binding would need more energy to overcome the binding and accordingly results in a larger redox potential. The detailed theory, as outlined in Scheme 2, has been shown elsewhere [22, 23]. Despite the reactions, the binding substrate in the electrochemical system would normally involve desorption, diffusion to the surface of electrodes, and the terminal redox process. Once the diffusion is eliminated with sonication, the desorbing behavior of the binding substrate is therefore directly correlated to the change of the redox potential. As such, the desorbing electrochemistry was performed in accordance with the paradigm. As shown in Fig. 11, at 25 °C, DNPO which bound to the SI layer showed the desorption-reduction potential at − 359 mV (cf. a). The potential was dramatically decreased to − 204 mV at 35 °C (cf. b) and there was no significant change at 45 °C (− 202 mV; cf. c). The SI layer demonstrated a stronger interaction at 25 °C than at 35 °C and at 45 °C. In contrast, the change of the desorption-reduction potential was reversed in the SVA layer, where the increasing temperature led to a dramatically-increased potential at 45 °C (− 831 mV; cf. f) and there was no essential difference available between 25 °C (− 201 mV; cf. d) and 35 °C (− 205 mV; cf. e). The SVA layer demonstrated a stronger interaction at 45 °C than at 25 °C and at 35 °C. Compared with the corresponding potentials in the SI layer (i.e., − 359 mV at 25 °C and − 202 mV at 45 °C), the desorption-reduction potential of DNPO in the SVA layer was lower at 25 °C (− 201 mV) but much higher at 45 °C (− 831 mV). In conjunction with the DLS spectra (cf. Fig. 8), these outcomes indicate that the catalytic channeling in this artificial reactor was temperature-dependent and alterable between the SI-layer and SVA-layer. The stronger interaction at 25 °C in the SI layer than in the SVA layer would allow the SI layer to preferentially “seize” DNPO. At 45 °C, the reverse was true between the SI-layer and SVA-layer, in which the SVA layer would preferentially capture DNPO. Hence, the alterable substrate channeling in the artificial reactor may be responsible for the occurrence of the cascade/non-cascade-switchable catalytic ability.

Schematic presentation of the desorbing electrochemical process with the binding molecule B

Reduction profiles with DNPO desorbing from two switchable outward layers in the novel reactor SVA-AmAg-SI (a SI layer at 25 °C; b SI layer at 35 °C; c SI layer at 45 °C; d SVA layer at 25 °C; e SVA layer at 35 °C; f SVA layer at 45 °C)
To further track this, Table 1 presents the desorption-reduction potentials for DNPO desorbing from all the prepared layers. With increasing temperature, the change of the desorption-reduction potentials in the non-responsive NT layer and AmAg layer was actually small and accordingly suggested no actual change in the interaction with DNPO. In contrast, the change of the desorption-reduction potentials in both the SI-layer and SVA-layer was much larger and accordingly exhibited the switchable behaviors in the two thermosensitive layers. The negative change in the SVA layer suggested an increasing interaction with DNPO (cf. Delta 2), in contrast to the positive change in the SI layer which revealed a decreasing interaction with DNPO (cf. Delta 1). In conjunction with the catalytic studies, these outcomes indicate again that the switchable substrate channeling was the reason behind the occurrence of the cascade/non-cascade-switchable catalytic ability.
4 Conclusions
This study met the present challenge in self-controlled catalysts by presenting an artificial polymer reactor capable of cascade/non-cascade-switchable catalytic ability. By seeking inspirations from nature, this polymer reactor was fabricated with tri-layer architectures that were composite of two inversely-thermosensitive outward layers and a non-responsive middle layer. The opposite thermosensitive responsiveness at the two outward layers induced switchable shapes in the resulting reactor, leading to cascade/non-cascade-alterable substrate channeling to the active middle layer. In this way, this artificial reactor led to the occurrence of cascade/non-cascade-switchable catalytic ability. It is therefore shown that functional catalysts capable of cascade/non-cascade-switchable catalytic ability can be realized by developing artificial reactors. The sustainable endeavors in the future will help increase the potential for applications and lead to the appearance of novel functional catalysts and catalytic materials.
References
T.M. Onn, M. Monai, S. Dai, E. Fonda, T. Montini, X. Pan, G.W. Graham, P. Fornasiero, R.J. Gorte, J. Am. Chem. Soc. 140, 4841 (2018)
S. Chen, J. Duan, P. Bian, Y. Tang, R. Zheng, S.Z. Qiao, Adv. Energy Mater. 5, n1500936 (2015)
M. Massaro, V. Schembri, V. Campisciano, G. Cavallaro, G. Lazzara, S. Milioto, R. Noto, F. Parisib, S. Riela, RSC Adv. 6, 55312 (2016)
Y.B. Huang, J. Liang, X.S. Wang, R. Cao, Chem. Soc. Rev. 46, 126 (2017)
T.L. Lohr, T.J. Marks, Nat. Chem. 7, 477 (2015)
J. Su, C. Xie, C. Chen, Y. Yu, G. Kennedy, G.A. Somorjai, P. Yang, J. Am. Chem. Soc. 138, 11568 (2016)
F. Rudroff, M.D. Mihovilovic, H. Gröger, R. Snajdrova, H. Iding, U.T. Bornscheuer, Nat. Catal. 1, 12 (2018)
A. Küchler, M. Yoshimoto, S. Luginbühl, F. Mavelli, P. Walde, Nat. Nanotechnol. 11, 409 (2016)
Z.J. Wang, C.N. Zhu, W. Hong, Z.L. Wu, Q. Zheng, Sci. Adv. 3, e1700348 (2017)
S. Li, S. Gong, J. Phys. Chem. B 113, 16501–16507 (2009)
C. Zuo, W. Wei, Q. Zhou, S. Wu, S. Li, ChemistrySelect 2, 6149 (2017)
Y. Han, X. Yuan, M. Zhu, S. Li, M. Whitcombe, S.A. Piletsky, Adv. Funct. Mater. 24, 4996 (2014)
P. Xiao, S. Wu, X. Shen, M. Zhu, S. Li, ChemCatChem 10, 5231 (2018)
R. Luo, M. Zhu, X. Yuan, S. Li, RSC Adv. 5, 5598 (2015)
J. Wang, M. Zhu, X. Shen, S. Li, Chem. Eur. J. 21, 7532 (2015)
R. Zhao, J. Hu, C. Niu, Y. Li, M. Hu, R. Liu, S. Li, J. Mater. Chem. C 4, 4748 (2016)
S. Li, Y. Luo, M.J. Whitcombe, S.A. Piletsky, J. Mater. Chem. A 1, 15102 (2013)
C.C. Tsai, G.F. Payne, J. Shen, Chem. Mater. 30, 8597 (2018)
T. Tripathy, H. Kolya, S. Jana, M. Senapati, Eur. Polym. J. 87, 113 (2017)
B. Singh, V. Sharma, Polymer 91, 50 (2016)
V. Amendola, Phys. Chem. Chem. Phys. 18, 2230 (2016)
W. Wei, M. Zhu, X. Shen, S. Wu, S. Li, RSC Adv. 6, 42869 (2016)
Y. Zhou, M. Zhu, S. Li, J. Mater. Chem. A 2, 6834 (2014)
Acknowledgements
The authors want to express their gratitude to the National Natural Science Foundation of China (Nos. 51473070 and 51808263). Thanks also should be expressed to the Jiangsu Province for support under the innovation-entrepreneurship program (Surencaiban [2015]26).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lu, Y., Wei, W., Zhu, M. et al. Polymer Reactor with Alterable Substrate Channeling for the Formation of Cascade/Non-cascade-Switchable Catalytic Ability. J Inorg Organomet Polym 30, 2039–2049 (2020). https://doi.org/10.1007/s10904-019-01349-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-019-01349-z