
Abstract
This paper describes the synthesis, as well the structural of a novel complex of Cu(II) with pyridine-2,6-dicarboxylic acid and melamine. The complex was characterized by differential scanning calorimetry (DSC) analysis, Fourier transform infrared and Raman spectroscopy with a detailed analysis of Hirshfeld surfaces and fingerprint plots. The single X-ray diffraction studies have revealed that the compound crystallizes in triclinic P\(\bar{1}\) space group with cell parameters a = 6.9839 (14), b = 11.037 (2), c = 12.990 (2), α = 72.268 (9), β = 75.237 (9), δ = 87.855 (10) and Z = 1. The crystal structure was stabilized by an extensive network of N–H···Br, N–H···O, O–H···N, O–H···Br, C–H···Br and non-classical N–H···N hydrogen bonds. Furthermore, the room temperature IR and Raman spectra of the title compound were recorded and analyzed on the basis of literature data. The DSC study shows three anomalies at 385, 495 and 507 K.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Pyridine-2,6-dicarboxylic acid (H2pdc) is an important chemical substance with obvious biological activity and has a wide application in medicine. In the past few years, much attention have been paid to the metal complexes involving H2pdc [1, 2] because it is found that the pyridine ring is more absorbable and has less side-effects than benzene ring. H2pdc is a water-soluble, commercially available, cheap and versatile N,O-chelator possessing diverse coordination modes, with a recognized biological function in the body metabolism [3], and in a variety of processes as an enzyme inhibitor [4], plant preservative [5] and food sanitizer [6]. In recent years, the H2pdc molecule has been used in combination with different metal transitions such as Zn, Fe, and Cu… to characterize a new complexes class containing organic and inorganic parts [7–9]. Melamine (M), a nitrogen rich compound is used in the manufacturing of plastics, whiteboards, kitchenware, etc. on the addition of formaldehyde. It is also used as a flame-resistant material due to content of excess of nitrogen [10]. High content of nitrogen confers the characteristics of protein molecules and hence it is being used to determine and maintaining the protein level in food and milk [11].
This article presents the synthesis of a Cu(II) complex with H2pdc and M by slow evaporation method at room temperature. The crystal structure has been determined by single-crystal X-ray diffraction. Their Hirshfeld surfaces analysis, infrared and Raman spectroscopic properties were also discussed. The differential scanning calorimetry (DSC) study was analyzed.
2 Experimental
2.1 Synthesis of the (H2pdcCuBr2)2∙2(MH+)∙2Br− Compound
The organic–inorganic hybrid compound, was prepared by the reaction of H2pdc (C7H5NO4), 1,3,5-triazine-2,4,6-triamine (C3H6N6) and copper (II) bromide (CuBr2) in concentrated HBr acid (38 %). The resulting solution was well stirred then kept at room temperature. After 2 weeks, dark purple crystals were formed. The slow evaporation allows the obtention of very high quality crystals of (H2pdcCuBr2)2∙2(MH+)∙2Br−.
2.2 X-Ray Collection
The twin crystal diffraction data was collected with a Bruker AXS CCD area detector system equipped with graphite monochromatic Mo Kα radiation (0.71073 Å) at room temperature. Data were collected at room temperature from an as-synthesized specimen measuring 0.33 × 0.27 × 0.20 mm3 in size. Atomic scattering factors were taken from the International Tables for X-ray crystallography [12]. There were 14,567 reflections collected in the whole Ewald sphere for 1.7 ≤ θ ≤ 25.9 of which 3593 reflections had an intensity of I > 2σ(I). The structure was solved by direct methods using SHELX-97 program incorporated in the OLEX2 program package [13, 14]. The hydrogen atoms of the cations were set in geometrical positions with a riding model. The final anisotropic full-matrix least squares refinement resulted in a convergence of the R factor at 0.034, Rw = 0.064. Selected crystallographic data and experimental condition details are presented in Table 1. The atomic coordinates and equivalent isotropic thermal agitation factors for atoms of the title compound are shown in Tables 2 and 3 respectively. The crystal structures and packing interactions are depicted using ORTEP3 [15] and DIAMOND [16] respectively.
2.3 Spectroscopic Measurement
Room temperature infrared absorption spectra in the 4000–400 cm−1 frequency range were recorded on a Nicole Impact 410 FT-IR spectrophotometer using standard KBr pellet technique with a spectral resolution of 2.0 cm−1. The pellets were prepared by mixing 15 mg of a powder sample with 300 mg of KBr. The heating of the (H2pdcCuBr2)2∙2(MH+)∙2Br− pellet in the temperature range 298–503 K; with a heating rate of 10 K min−1, was performed in an air atmosphere Spectra heating cell. A thermocouple Eurotherm regulator was used for the temperature measurements. As for the Raman spectra, they were performed at room temperature using a LABRAMHR 800 triple monochromatic instrument. The excitation line was 514.5 nm. The laser beam was focused on the sample through a ×50 Microscope Objective and the laser spot dimensions were around 10 μm2. Concerning the laser power, it was kept <5 mW so as to avoid the sample heating.
2.4 Calorimetric Measurements
The DSC measurements were recorded on a NETZSCH apparatus (Model 204 Phoenix) at a heating rate of 5 K min−1 at the temperature range from 300 to 551 K.
3 Results
3.1 Structure Description
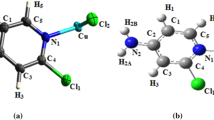
The title compound crystallizes in triclinic crystal system, P\(\bar{1}\) (Ci in Schönflies notation) space group with one formula units in unit cell (Z = 1). The ORTEP diagram with the numbering scheme is shown in Fig. 1. The asymmetric unit contain a half of independent [H2pdcCuBr2], one cation (MH+) and an isolated bromide ion Br−. The molecular structure of (H2pdcCuBr2)2∙2(MH+)∙2Br− is shown in Fig. 2. The crystal structure is organized in layers staked along [011] direction. The (H2pdcCuBr2) are located on planes perpendicular to the a axis at z = 0, and those for organic groups (MH+) are developed along the b axis at z = ½. Between them are interposed two free bromine atoms Br and two water molecules. The Main distances (Å) and the bond angles (º) in the (H2pdcCuBr2)2.2(MH+).2Br− crystals were assembled in Table 4. The Cu atom has lightly distorted octahedral coordination geometry, defined by one Nitrogen and two carboxylate oxygen atoms from pdc (O1, O4) and one bromine atom in the equatorial plane while two bromine atoms coordinated occupying the axial positions. The bond distance for Cu1–N7 of 2.017 (3) Å is similar to those of related Cu(II) complexes [17–19]. The bond length of Cu1–O2 and Cu1–O3 are 2.501 (3) Å and 2.394 (3) Å respectively. The bond lengths and angles are comparable with those for the related Cu(II) compounds [20]. The protonation of N2 leads the increase in C1–N2 and C2–N2 bond lengths than that of other ring C–N bonds. The protonation of melamine affect the side chain C1–N1, C2–N3 and C3–N5 bond lengths. The internal C1–N2–C2 angle (119.6 (4)) at protanated N atom is significantly greater than other two angles C2–N4–C3 (116.9 (4)) and C1–N6–C3 (115.6 (4)) within the ring. It is worth while mentioning, that in recently determined structure of melaminium hydrogenphthalate [21], melaminium chloride hemihydrates [22] and bis(melaminium) sulphate dehydrate [23], the melaminium residues exhibit similar pattern of the internal C–N–C and N–C–N angles. As a result of the protonation of the melamine ring the internal angle N–C–N containing only non-protonated nitrogen atom N4–C3–N6 (125.5 (4)) is significantly greater than both N–C–N angle containing protonated nitrogen atoms N2–C2–N4 (120.2 (4)) and N2–C1–N6 (122 (4)). Figure 3 shows a perspective view of the crystal structure, together with the network of hydrogen contacts (dotted lines). There are several types of hydrogen bonding interactions present in the crystal structure. The classic bonds of the type N–H···Br are between the hydrogen of the N2 and the N3 and the isolated bromides Br−. Overall, the crystal structure is stabilized via the various types of hydrogen bonding interactions N–H···O, N–H···Br–Cu, O–H···N, C–H···Br and O–H···Br. The organic groups are interconnected by non equivalents N–H···N hydrogen bonds. These hydrogen bonds formed of rings, according to the graph theory these two rings are denoted; \({\text{R}}_{2}^{2}\) (8) [24]. The hydrogen-bonding data are summarized in Table 5.

An ORTEP view of (H2pdcCuBr2)2∙2(MH+)∙2Br− with displacement ellipsoids drawn at the 50 % probability level. Symmetry codes: (i) −x + 1, −y + 1, −z

Crystal packing diagram of (H2pdcCuBr2)2∙2(MH+)∙2Br−

The drawing show the intermolecular hydrogen bonds contacts which are represented by dotted line for (H2pdcCuBr2)2∙2(MH+)∙2Br− Symmetry codes: (ii) x + 1, y, z;(iii) −x + 1, −y + 1, −z + 1; (iv) x, y, z + 1; (v) x, y, z−1; (vi) −x + 2, −y, −z + 1; (vii) −x + 1, −y, −z + 1
3.2 IR and Raman Spectroscopy
Although IR spectroscopy is one of the major physical methods of investigation of molecular structure, we have studied the IR spectra of (H2pdcCuBr2)2∙2(MH+)∙2Br−. The assignment of the vibrational bands was performed by comparison with the vibration modes frequencies of homologous compounds [25–27]. Figure 4 shows the superposition of the IR and Raman spectrum of the reported compound performed at room temperature between 4000–400 and 4000–50 cm−1 respectively. The assignments of the bands observed in the IR and Raman spectra of (H2pdcCuBr2)2∙2(MH+)∙2Br− are listed in Table 6. It is to be noted that the high-frequency region 3605 and 3535 cm−1 in IR is related to the asymmetric and symmetric stretching of (OH). Furthermore, the bands observed at 3314 and 3074 cm−1 in IR respectively, is ascribed to asymmetric and symmetric stretching of NH2. The bands attributed to the (N–H) and (C–H) stretching modes are experimentally observed from 2881 to 2448 cm−1 in the IR spectrum. The (N–H) deformation mode of the title compound was observed at 1717 cm−1. Those observed at 1658 and 1584 cm−1 in IR and at 1581 cm−1 in Raman, respectively, are assigned to the asymmetric and symmetric bending of NH2. The side chain C–N stretching bands are assigned at 1522 cm−1 in IR. The IR bands observed at 1478 cm−1, is accredited to the (O–H) deformation mode. The C=C stretching mode is exhibited at 1374 cm−1 in IR and 1378 cm−1 in Raman. The observed bands at 1227, 1175 cm−1 are associated with the (C=O) stretching mode. The NH2 rocking bands are observed at 1085 and 1025 cm−1 in IR spectrum. The C–N–C and N–C–N out of plane bending is assigned at 768 and 690 cm−1. The in-plane C–N–C and N–C–N bands are assigned at 727 and 629 cm−1 respectively. Furthermore, the (C–N–C) and the (C–C–C) deformation are observed at 539 cm−1. The weak bonds at 501 and 408 cm−1 are from the Cu–N and Cu–O vibrations, respectively. The bands observed at 60 and 118 cm−1 are assigned to Br–Cu–Br bending. The Cu–Br asymmetric stretching mode is found at 249 and cm−1. The symmetric stretching of Cu–Br appeared between 197 and 168 cm−1.

Superposition of FT-IR and the Raman spectra of (H2pdcCuBr2)2∙2(MH+)∙2Br−compound at room temperature
3.3 Thermal Properties
The thermogram of (H2pdcCuBr2)2∙2(MH+)∙2Br− crystals shows the existence of three endothermic peaks located at 385, 495 and 507 K (Fig. 5). The measured enthalpy, ΔH1, for the first peak which corresponds to dehydration of the water molecule is 1.145 kJ g−1. The second and third located at around 495 and 507 K corresponds in the phase transition anomaly. These two peaks overlap and, consequently, the enthalpy for the sum of these peaks, ΔH2 = 1.417 kJ g−1.

DSC curves of (H2pdcCuBr2)2∙2(MH+)∙2Br−
Figure 6 shows the superposition of IR spectra and the zoom of the zone 4000–400 cm−1 in the temperature range 298–503 K. A progressive variation of the speed of spectra was detected in the zone characterizing the vibrations of strain of the water; this variation is observed between the temperatures 353 and 383 K. We also report the displacement of certain peaks and the disappearance of the others in the field along the low-frequency, which indicates the departure of an element of the structure. This study is in agreement with the results that were found by the previous thermal study the DSC.

FT-IR spectra at different temperatures between 298 and 503 K for (H2pdcCuBr2)2∙2(MH+)∙2Br−
3.4 Hirshfeld Surfaces
The Hirshfeld surfaces [28–38] and the associated 2D fingerprint plots [39–43] calculated using Crystal Explorer [44] which accepts a structure input file in the CIF format. The Hirshfeld surfaces enclosing a molecule are defined by points where the contribution to the electron density from the molecule of interest is equal to the contribution from all the other molecules. The Hirshfeld surfaces dnorm maps, of the (H2pdcCuBr2)2 are illustrated in Fig. 7. The 2D fingerprint plots of (H2pdcCuBr2)2 are shown in Fig. 8. They clearly show the influences of different co-formers on the intermolecular interactions of the (H2pdcCuBr2)2. The large and deep red spots on the dnorm Hirshfeld surfaces indicate the close-contact interactions, which are mainly responsible for the significant hydrogen bonding contacts. The 2D fingerprint plots are displayed by using the expanded 0.6–2.8 Å view with the de and di distance scales on the graph axes. In the fingerprint region (Fig. 8), H···Br (6.4 %) interactions are represented by a spike in the top area whereas the Br···H (17.3 %) interactions are represented by a spike in the lower part of fingerprint plot. Hydrogen bonding interactions H···H (17.3 %) are very high compared to the other bonding interactions. The 2D fingerprint plot reveals that the O···H/H···O interaction covers 22.6 % of the total surface. Additionally, the C···H/H···C (12.5 %) and N···H/H···N (5.5 %) short contacts have contributed in the 2D fingerprint plot. The Br···Br (3.6 %) molecular interactions appear as a tower like shape at the middle part of the fingerprint plot. The Hirshfeld surfaces analysis shows the existence of other weak intermolecular contacts, for which the percentage participations to the Hirshfeld surfaces area are presented in Fig. 9.

Hirshfeld surfaces mapped with dnorm of (H2pdcCuBr2)2

Fingerprint plot of (H2pdcCuBr2)2

Relative contribution of various intermolecular interactions to the Hirshfeld surface area
The Hirshfeld surfaces dnorm of the (MH+) is illustrated in Fig. 10. In the fingerprint region (Fig. 11), the amount of H···H interactions comprises 23.8 % of the Hirshfeld surfaces. The N···H/H···N intermolecular interactions appear as two distinct spikes in the 2D fingerprint plots, labeled correspondingly. The N···H/H···N hydrogen bonding intermolecular interactions has 22.2 % contribution to the total Hirshfeld surfaces. While O···H/H···O hydrogen bonding interactions only comprises 19.5 % of the total Hirshfeld surfaces. The proportion of Br···H/H···Br interactions comprises 13.3 % of the Hirshfeld surfaces for molecule (MH+). The percentages of them are summarized in Fig. 12.

Hirshfeld surfaces mapped with dnorm of (MH+)

Fingerprint plot of (MH+)

Percentage contributions to the Hirshfeld surface area for various contacts
4 Conclusion
New hybrid compound (H2pdcCuBr2)2∙2(MH+)∙2Br− were grown by the slow solvent evaporation solution growth technique at ambient temperature. The crystal structure is stabilized via the various types of hydrogen bonding interactions. The assignment of the vibrational bands was performed by comparison with the vibration modes frequencies of homologous compounds. The DSC thermal analysis was performed to establish the thermal stability of the crystal. The findings from DSC analyses revealed the existence of two-phase transitions at 495 and 507 K. Hirshfeld surfaces display all of the intermolecular interactions within the crystal at once and are therefore ideal for analyzing the crystal packing. Hirshfeld surfaces were employed to confirm the existence of intermolecular interactions in (H2pdcCuBr2)2 and (MH+).
5 Supplementary Material
Supplementary crystallographic data for this article in CIF format are available as Electronic Supplementary Publication from Cambridge Crystallographic Data Centre (CCDC 1401522). This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html, or from the Cambridge Crystallographic Data Centre, 12 Union Rood, Cambridge CB2 1EZ, UK (Fax: (international): +44 1223/336 033; e-mail: deposit@ccdc.cam.ac.uk).
References
D.P. Murtha, R.A. Walton, Metal carboxylates. VI. mixed ligand complexes of copper (II) containing pyridine-2,6-dicarboxylic acid in its monoanionic and dianionic forms. Inorg. Chim. Acta 8, 279–284 (1974)
X.X. Zhong, X.L. Wang, Y.M. Jiang, Crystal structure and synthesis of cobalt pyridine 2,6-dicarboxylate. J. Guangxi Normal Univ. 22, 60 (2004)
T. Douki, B. Setlow, P. Setlow, J. Photochem. Photobiol. B Biol. 4, 591–597 (2005)
K. Murakami, Y. Tanemura, M. Yoshino, J. Nutr. Biochem. 14, 99–103 (2003)
Y. Kazuhiro, Y. Noriko, F. Tadayasu, Eur. Patent EP0603165, 591–597 (1994)
G.A. Burdock, Encyclopedia of Food and Color Additives, CRC Press, New York (1996)
F.A. La Porta, P.H. Ramos, E.C. de Resende, M.C. Guerreiro, J.O.S. Giacoppo, T.C. Ramalho, J.R. Sambrano, J. Andrés, E. Longo, J. Inorg. Chem. Acta 416, 200–206 (2014)
G.A. van Albada, M. Ghazzali, K. Al-Farhan, E. Bouwman, J. Reedijk, J. Polyhedron. 52, 1059–1064 (2013)
M. Brandi-Blanco, D. Choquesillo-Lazarte, A. Domínguez-Martín, A. Matilla-Hernández, J. González-Pérez, A. Castiñeiras, J.M. Niclós-Gutiérrez, J. Inorg. Biochem. 127, 211–219 (2013)
Y. Tyan, M. Yang, S. Jong, C. Wang, J. Shiea, Anal. Bioanal. Chem. 395, 729–735 (2009)
E.L. Kennaway, Biochem. J. 15, 510–512 (1921)
C. Kluwer, International Tables for X-ray crystallography, Dordrecht (1992)
G.M. Sheldrick, Acta Crystallogr. Sect. A 64, 112–122 (2007)
O.V. Dolomanov, L.J. Bourhis, R.J. Gildea, J.A.K. Howard, H. Puschmann, J. Appl. Cryst. 42, 339–341 (2009)
L.J. Farrugia, ORTEP3 for windows. J. Appl. Crystallogr. 30, 565 (1997)
K. Brandenburg, (1998) Diamond Version 2.0 Impact Gbr. Bonn, Germany
M. Koman, M. Melník, M. Moncol, Inorg. Chem. Commun. 3, 262–266 (2000)
J. Shou-Wen, C. Wan-Zhi, Polyhedron 26, 3074–3084 (2007)
C. Julienne, M. Jerome, R. Didier, Acta Crystallogr. Sect. C 60, m101–m103 (2004)
P.K. Bhaumik, S. Jana, S. Chattopadhyay, Inorg. Chim. Acta 390, 167–177 (2012)
J. Janczak, G.J. Perpetuo, Acta Cryst. C 57, 123–125 (2001)
J. Janczak, G.J. Perpetuo, Acta Crystallogr. C 57, 1120–1122 (2001)
J. Janczak, G.J. Perpetuo, Acta Crystallogr. C 57, 1431–1433 (2001)
J. Bernstein, R.E. Davis, L. Shimoni, N.-L. Chang, J. Angew, Chem. Int. Ed. Engl. 34, 1555–1573 (1995)
N. Karâa, B. Hamdi, A. Ben Salah, R. Zouari, J. Mol. Struct. 1013, 48–58 (2012)
C. Ben Nasr, I. Said, M. Rzaigui, Mater. Res. Bull. 36, 789–798 (2001)
N. Chaari, Z. Abdelkefi, S. Chaabouni, N. Chniba-Boudjada, P. Bordet, Phase Transit. 81, 101–111 (2008)
M.A. Spackman, J.J. McKinnon, CrystEngComm 4, 378–392 (2002)
F.P.A. Fabbiani, C.K. Leech, K. Shankland, A. Johnston, P. Fernandes, A.J. Florence, N. Shankland, Acta Cryst. C 63, o659–o663 (2007)
L. Checinska, S. Grabowsky, M. Malecka, A.J. Rybarczyk-Pirek, A. Józwiak, C. Paulmann, P. Luger, Acta Cryst. B 67, 569–581 (2011)
H. Pérez, R.S. Corrêa, A.M. Plutín, B. O’Reilly, M.B. Andrade, Acta Cryst. C. 68, o19–o22 (2012)
S.K. Seth, D. Sarkar, A.D. Jana, T. Kar, Cryst. Growth Des. 11, 4837–4849 (2011)
S.K. Seth, I. Saha, C. Estarellas, A. Frontera, T. Kar, S. Mukhopadhyay, Cryst. Growth Des. 11, 3250–3265 (2011)
S.K. Seth, D. Sarkar, T. Kar, CrystEngComm 13, 4528–4535 (2011)
S.K. Seth, D. Sarkar, A. Roy, T. Kar, CrystEngComm 13, 6728–6741 (2011)
S.K. Seth, N.C. Saha, S. Ghosh, T. Kar, Chem. Phys. Lett. 506, 309–314 (2011)
S.K. Seth, P.C. Mandal, T. Kar, S. Mukhopadhyay, J. Mol. Struct. 994, 109–116 (2011)
P. Manna, S.K. Seth, A. Das, J. Hemming, R. Prendergast, M. Helliwell, S.R. Choudhury, A. Frontera, S. Mukhopadhyay, Inorg. Chem. 51, 3557–3571 (2012)
S.K. Seth, G.C. Maity, T. Kar, J. Mol. Struct. 1000, 120–126 (2011)
J.J. McKinnon, M.A. Spackman, A.S. Mitchell, Acta Crystallogr. Sect. B 60, 627–668 (2004)
A.L. Rohl, M. Moret, W. Kaminsky, K. Claborn, J.J. Mckinnon, B. Kahr, Cryst. Growth Des. 8, 4517–4525 (2008)
A. Parkin, G. Barr, W. Dong, C.J. Gilmore, D. Jayatilaka, J.J. Mckinnon, M.A. Spackman, C.C. Wilson, CrystEngComm 9, 648–652 (2007)
F.P.A. Fabbiani, L.T. Byrne, J.J. Mckinnon, M.A. Spackman, CrystEngComm 9, 728–731 (2007)
S.K. Wolff, D.J. Grimwood, J.J. McKinnon, D. Jayatilaka, M.A. Spackman, in Crystal Explorer 2.1, University of Western Australia, Perth, 2007
Acknowledgments
The authors thank the members of units of common services, at the University of Sfax for their assistance in the measurements for X-ray diffraction. The authors are also thankful to Prof Hamadi KHEMAKHEM, for his co-operating in the Raman spectroscopy measurement.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mesbeh, R., Hamdi, B. & Zouari, R. (H2pdcCuBr2)2∙2(MH+)∙2Br− (M = Melamine, H2pdc = Pyridine-2,6-Dicarboxylic Acid): Crystal Structure, Hirshfeld Surface Analysis, Vibrational and Thermal Studies. J Inorg Organomet Polym 26, 744–755 (2016). https://doi.org/10.1007/s10904-016-0371-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10904-016-0371-1