
Abstract
The structural, electronic and photophysical properties of three new asymmetric, highly fluorescent difluoroborondipyrromethene (BODIPY) dyes, bearing an amino or an acetamido group at position 3 of the chromophoric core, have been studied in different apolar, polar and polar/protic solvents. The presence of the 3-amido group extents the delocalization of the π-system, leading to bathochromic shifts in the absorption and fluorescence bands, as predicted by quantum mechanic calculations. The 3-amino dye shows photophysical properties highly dependent on the solvent polarity and acidity, and is characterized by a hypsochromic shift of its absorption band, with regard to the corresponding acetylated dye, as well as a low fluorescence quantum yield in acid media with proton concentration lower than 4 × 10−4 M. In media with higher proton concentration, the BF2 bridge group of the 3-amino dye is removed, yielding the corresponding non-fluorescent dipyrromethene precursor. These results suggest that the 3-amino dye could be used as a fluorescence probe for the study of the acidity of different environments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Borondipyrromethene dyes (BODIPY dyes) show interesting photophysical and lasing properties [1–8]. In general, they present strong absorption and fluorescence bands in the visible region and high fluorescence quantum yields, in many cases close to the unity, depending on their structure and the environmental conditions [8]. BODIPY dyes have been also used as active media for tunable dye lasers with high energy conversion outputs. These high laser efficiencies are mainly due to a high radiative deactivation probability, a low non-radiative deactivation rate constant, a low triplet–triplet absorption and a high photostability [9–12]. BODIPY dyes have been also widely applied as fluorescent sensors and probes to study biological systems containing lipids, nucleic acids or proteins [13–15], as well as light harvesting arrays to develop antenna systems [16–19].
In previous works, it has been demonstrated that the photophysical properties of the BODIPY dyes can be modulated to some extent by the incorporation of suitable substituents in the chromophoric core of these dyes [20–24]. In this sense, theoretical simulations are powerful tools to predict the photophysical behavior of new analogs and to guide the synthesis of new molecular structures with specific physicochemical properties [25, 26]. For instance, the incorporation of electron-donor or electron-withdrawing substituents can induce the formation of intra- or inter-molecular charge transfer complexes, making the photophysical properties of the dyes strongly sensitive to environmental factors. These are fundamental aspects for the applications of BODIPY dyes as molecular probes [27–29].
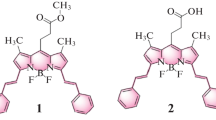
In the present work, the photophysical properties of three new BODIPY dyes, bearing a 3-amino (dye 1a) or 3-acetamido (dyes 2a and 2b) group at position 3 (Scheme 1), have been studied in liquid solution. These asymmetric dyes were synthesized by a non-conventional route described elsewhere [30]. The photophysical properties of these dyes have been studied in six representative solvents, ranging from apolar (cyclohexane) to polar (acetone and ethyl acetate) and polar/protic (ethanol, methanol and 2,2,2-trifluoroethanol) solvents. The effects of other factors, such as the dye concentration and the medium acidity, have been also considered. The photophysical study has been complemented with quantum mechanic calculations in which the geometry, electronic distribution and spectral properties of these dyes have been analyzed.

Molecular structure of the dyes 1a, 2a and 2b
Experimental
The synthesis and structural elucidation of the dyes have been previously described [30]. All solvents were of spectroscopic grade (Merck, Aldrich) and were used without further purification. Dye solutions of different concentrations were prepared from a stock solution (1.5 × 10−3 M) of the corresponding dye in acetone, after vacuum evaporation of the solvent and adequate dissolution of the residue. Diluted dye solutions in ethanol were acidified or basified by adding adequate drops of HCl 7 M in ethanol (prepared by bubbling HCl gas through ethanol up to saturation) and NaOH 1 M in the same solvent, respectively.
UV-Vis absorption and fluorescence spectra were recorded on a Varian Cary 4E spectrophotometer and on a SPEX Fluorolog 3-22 spectrofluorimeter, respectively, using quartz cells of different optical pathlengths (from 1 to 0.001 cm), depending on the dye concentration. Fluorescence spectra were corrected from the emission monochromator wavelength dependence and the photomultiplier sensibility. The fluorescence spectra of diluted samples were registered in the right-angle configuration, while the front-face mode was applied for solutions with high optical densities to minimize reabsorption and reemission phenomena. In the last case, the sample was oriented 0° and 22.5° with respect to the excitation and emission beams, respectively. Fluorescence quantum yields (Φ) were evaluated using reference samples emitting at the same spectral region: a diluted solution of PM567 in methanol (Φ r = 0.91) [31] for the 3-amino dye 1a (excitation at 490 nm) and a diluted solution of PM605 in ethanol (Φ r = 0.65) [20] for the 3-acetamido dyes 2a and 2b (excitation at 510 nm).
Radiative decay curves were registered with the time corelated single-photon counting technique (Edinburgh Instruments, FL920). Emission was monitored at the maximal emission wavelength, exciting at 410 nm by means of a diode laser (PicoQuant, LDH410) with 150 ps FWHM pulses, 10 MHz repetition rate and a power supply of 0.65 mW. The fluorescence decay curves, deconvoluted from the instrument response function detected by a Ludox scatter suspension, were generally analyzed as monoexponentials [statistical parameters: chi-square χ 2 < 1.2 and Durbin–Watson (D.W.) >1.75], except in specified cases, from which the fluorescence lifetime (τ) was obtained. The experimental errors in the determination of Φ and τ values were estimated to be 5% and 1%, respectively.
HPLC analysis of the HCl-induced change 2a→3a was carried out at room temperature in a diode-array Agilent 1100 apparatus equipped with a C18 (5 μm) reverse phase column, 4.6 × 150 mm, with acetonitrile–water 9:1 v/v as eluent at a flow rate of 2 ml min−1. Under these conditions, the retention times were 1.36 min (2a, λ max 490 nm) and 0.86 min (3a, λ max 460 nm).
Quantum mechanic calculations were carried out using the Gaussian 03 software [32]. The ground state geometry was optimized by the Density Functional Theory (DFT) using the hybrid B3LYP method and the double valence (6–31G) basis set. The first excited state was optimized with the Configuration Interaction Singles (CIS) method with the same basis set. The absorption and fluorescence energy gaps were estimated by the S0–S1 Franck–Condon transition from the optimized geometry at the ground (S0) and first excited singlet (S1) states, respectively, using the Time-Dependent (TD-B3LYP) method. The solvent effect was simulated by means of the Polarizable Continuum Model (PCM).
Results and discussion
Figure 1 shows the absorption and fluorescence spectra and the decay curves of diluted solutions of the dyes 1a and 2a in cyclohexane. The presence of the 3-acetamido group in 2a induces a bathochromic shift of ca. 20 nm in the absorption and fluorescence spectral bands, with respect to those of 1a (Table 1). Dye 1a presents absorption and fluorescence bands in positions close to those of the model dye PM567, the symmetric commercial laser dye with alkyl substituents [8]. The shape of these spectral bands is similar to those of other described alkyl-BODIPY dyes [8], and, consequently, it can be deduced that the presence of a 3-amino or a 3-acetamido group in the BODIPY chromophoric core does not significantly alter the photophysics of BODIPY dyes, at least, in non-polar media.

Absorption (bold curves) and fluorescence-height normalized spectra (a) and fluorescence decay curves (b) of 2 × 10−6 M solutions of the dyes 1a (a) and 2a (b) in cyclohexane
The spectral shifts are theoretically predicted by quantum mechanic calculations. Indeed, the S0–S1 energy gap in both absorption and emission transitions of the 3-amino dye 1a is displaced around 0.10 eV at higher energies with respect to those of the 3-acetamido dye 2a. These results confirm previous conclusions, in the sense that the TD-B3LYP method is a valid tool to simulate the spectral bands of BODIPY dyes and to simulate molecular structures of similar dyes with specified absorption and fluorescence bands [25, 26]. The bathochromic shift of 2a can be ascribed to an augmentation in the delocalization of the electronic π-system of the BODIPY core, which is extended through the 3-acetamido group. Indeed, the contour maps of the HOMO and LUMO states (Fig. 2), reveal an extended delocalization of the electronic π-system to the amide substituent, explaining the observed bathochromic shift.

HOMO and LUMO electronic density of the dyes 1a and 2a calculated by the B3LYP/6–31G method
The S0–S1 transition probability, described by the oscillator strength (f) in absorption and by the radiative rate constant (k fl) in fluorescence, does not show any clear evolution with the substituent (Table 1). The experimental f value is lower for 1a, whereas the opposite behavior is observed in the experimental k fl value. This last evolution is predicted by theoretical k fl data, although similar theoretical f values are also proposed. Anyway, dye 1a has a higher non-radiative deactivation probability, which reduces its experimental fluorescence quantum yield and lifetime (Table 1).
On the other hand, the presence of tert-butyl groups at positions 1 and 6 in the 3-acetamido dye 2b increases the absorption (ɛ and f) and fluorescence (k fl) transition probability (Table 1), with regard to the presence of ethyl groups in 2a. This effect could be ascribed to the higher inductive effect of the tert-butyl substituent. Theoretical calculations also show this tendency (Table 1). The higher k fl value and the lower k nr value of the dye 2b with respect to 2a induce a higher fluorescent ability in 2b (Table 1).
In a previous work, it has been demonstrated that in the commercial PM597 dye the presence of bulky tert-butyl groups at positions 2 and 6 induces an important decrease in the Φ value due to an increase in non-radiative deactivation probability [33]. This increase in internal conversion processes has been ascribed to a distortion from the planarity in the S1 state induced by a sterical hindrance of the tert-butyl substituents with the adjacent methyl groups, as suggested by quantum mechanic calculations. However, in the case of the dye 2b such an effect must be much lower because the absence of substituent at the adjacent 8 position must allow the steric accommodation of the tert-butyl group at position 1, without altering the planarity of the rings of the aromatic chromophore. This is an important fact, because rigid and planar structures imply lower k nr values and, hence, more fluorescent systems [34].
The photophysical properties of the dyes have been registered in six representative solvents, including apolar, polar and protic solvents (Table 2). The spectral bands are hypsochromically shifted, as confirmed by theoretical calculations, and the fluorescence lifetime increases with the increase of the solvent polarity. This is a general behavior in BODIPY dyes [8]. However, the fluorescence quantum yield of the three studied dyes is nearly solvent independent.
Anyway, the most striking feature of the solvent effect is the drastic change in the shape of the absorption band in polar/protic media, mainly for the dye 1a (Fig. 3). The shape of its absorption band in cyclohexane reminds that of other alkyl substituted dyes of the same family, with a well-defined vibrational structure. By increasing the solvent polarity, the absorption band becomes broader and less intense, losing its vibrational structure. Thus, in polar/protic solvents the band is very wide and appears strongly shifted to higher energies. However, the fluorescence band reminds that of other BODIPY dyes (Fig. 3), with a large Stokes shift (1,150 cm−1 for dye 1a in methanol). Besides, the ɛ value at the absorption maximum strongly decreases with the solvent polarity, but this decrease is compensated by the broadening of the band, leading to an oscillator strength (area under the absorption curve) nearly solvent independent (Table 2).

Absorption (bold curves) and fluorescence spectra of 2 × 10−6 M solutions of 1a in cyclohexane (a), ethyl acetate (b), and 2,2,2-trifluoroethanol (c)
It has been previously observed a broadening of the absorption band in 8-p-phenylene-BODIPY dyes in polar solvents, with regard to non-polar ones [6]. This change has been assigned to the overlapping of the S0–S2 absorption band with the low-lying S0–S1 band. This S0–S2 transition has a charge transfer nature and is shifted to lower energies by increasing the solvent polarity, in such a way that in polar/protic solvents this absorption band completely overlaps with the S0–S1 absorption band. In this sense, it has been also observed a weak emission band at lower energies for an 8-aniline-BODIPY dye [35]. This emission has been ascribed to the fluorescence from an excited charge transfer state between the aniline group and the BODIPY core. According to these interpretations, charge transfer states should be also possible in the dyes herein studied between the 3-amino or 3-acetamido group and the electronic π-system of the chromophore core. This charge transfer state, which explains the observed broadening of the absorption band, should be non fluorescent, since the shape of the fluorescence band of these dyes is solvent independent (Fig. 3).
The charge transfer state should be affected by specific interactions between the 3-amino or 3-acetamido groups and protic solvents. This situation is very common for many amino-aromatic dyes in alcohols and has been widely studied by some of the authors for 7-amino-coumarine or rhodamine laser dyes [36, 37]. An electron transfer process from the amino group to the electronic π-system must be affected by hydrogen-bonding interactions between the NH2 group and the OH group of protic solvents. In the case of the 3-acetamido dyes 2a and 2b, these interactions would be less important because the N-atom is less basic, as confirmed by quantum mechanic calculations: the electron CHelpg charge at the N-atom is −0.88 for dye 1a and −0.65 for dye 2a.
Since the most significant changes in the photophysics of these dyes are observed in polar/protic media, the effect of the presence of acids in the photophysics of dyes 1a and 2a in solution is now considered. The absorption and fluorescence spectral properties of 2a are insensitive to the H+ and OH− concentration, except in extreme acid solutions as is shown in Fig. 4. Indeed, at high proton concentrations ([H+] >8 ×10−2 M) a new absorption band appears at higher energies, at around 470 nm. The fluorescence quantum yield decreases (from 0.70 to 0.36), while the fluorescence lifetime remains unaltered. Furthermore, the fluorescence quantum yield further decreases (Φ = 0.15) after excitation at this new absorption band. These results suggest the presence of a static quenching in acid media. Anyway, BODIPY dyes such as the commercial PM567 dye are chemically unstable in high acidity media [38].

Absorption (a) and fluorescence (b, scaled to the Φ value) spectra of the dye 2a in 2 × 10−6 M solutions in acid and basic ethanol: a [OH−] = 3 × 10−3 M; b [OH−] = 2 × 10−5 M; c [OH−] = 7 × 10−6 M; d [H+] = 3 × 10−8 M; e [H+] = 3 × 10−6 M; f [H+] = 4 × 10−5 M; g [H+] = 4 × 10−4 M; h [H+] = 2 × 10−3 M; i [H+] = 7 × 10−3 M; j [H+] = 5 × 10−2 M; k [H+] = 8 × 10−2 M
On the other hand, the 3-amino dye 1a shows important changes in the absorption and fluorescence spectra with the proton concentration in ethanolic solutions (Fig. 5). The absorption intensity progressively decreases by decreasing [OH−], without any appreciable change in the shape of the absorption band (Fig. 5a, curves a–c). In solutions with [H+] in the range 3 × 10−8 to 4 × 10−4 M (Fig. 5a, curves d–g), the absorbance at the vibronic shoulder increases in detriment of the main absorption band, and at [H+] values higher than 2 × 10−3 M a new hypsochromic band appears at 460 nm (Fig. 5a, curves h–l). These changes correlates with the modifications observed in the shape of the absorption band of 1a in 2,2,2-trifluoroethanol, where the absorption band is hypsochromically shifted and becomes broader (Fig. 3, curve c).

Absorption (a) and fluorescence (b, scaled to the Φ value) spectra of the dye 1a in 2 × 10−6 M solutions in acid and basic ethanol: a [OH−] = 3 × 10−3 M; b [OH−] = 2 × 10−5 M; c [OH−] = 7 × 10−6 M; d [H+] = 3 × 10−8 M; e [H+] = 3 × 10−6 M; f [H+] = 4 × 10−5 M; g [H+] = 4 × 10−4 M; h [H+] = 2 × 10−3 M; i [H+] = 7 × 10−3 M; j [H+] = 3 × 10−2 M; k [H+] = 5 × 10−2 M; l [H+] = 8 × 10−2 M
The shape and position of the fluorescent band, as well as the fluorescence quantum yield, of the dye 1a remind unaltered in basic media and at moderated acid environments (Fig. 5b, curves a–g). In high acidity media, [H+] >2 × 10−3 M, and after excitation at the new absorption band centered at 460 nm, the fluorescence band becomes very broad and its intensity drastically decreases (Fig. 5b, curves h–l). Consequently, the excitation at this absorption band in these acid media is characterized by a very low or null fluorescence quantum yield (Φ < 0.1), corresponding to the appearance of the non-fluorescent compound 3a, as is discussed below. The fluorescence decay curves cannot be further analyzed as monoexponential, and it is needed to consider up to three exponentials.
The acidity effect in the photophysics of 1a does not show the typical acid–base equilibrium between two molecular forms, since a return to neutral solutions did not reach the expected absorption and fluorescence bands. Moreover, the appearance of the hypsochromic absorption band, observed in acid solutions immediately after sample preparation, is also observed in aged solutions with moderated H+ concentrations. All these results support the previously described degradation of 1a in acid media, giving rise to the corresponding dipyrromethene precursor 3a through the loss of the BF2 unit (Scheme 2) [30]. This process should take place via the protonation of the NH2 group in 1a, much easier in this dye than in 2a. This reaction (Scheme 2), previously observed through the isolation and analysis of the product 3a [30], is now confirmed by HPLC analysis. This reaction must be favored in the amine-BODIPY dye 1a since the N atom is more basic in this derivative than in 2a. The loss of the BF2 bridge disrupts the chromophoric π-system, leading to a decrease of the aromaticity of the system and to a new absorption band at higher energies. Besides, the two pyrrole units in 3a could rotate in the excited state around the bonds connecting them [39] and, consequently, the compound is not fluorescent. Moreover, 3a must present a strong intramolecular hydrogen bond between the two nitrogen atoms in both possible proton-tautomeric structures (Scheme 2), and the proton transference in the excited state cannot be discarded. These processes could give rise to an extra loss of the excitation energy and, hence, to a further decrease of the fluorescent ability of 3a. In addition, the NH2 substituent could also acts as a proton donor group, increasing in this way the intramolecular proton transfer probability. All these phenomena can explain the poor fluorescent ability of 3a.

Generation of the dipirromethene compound 3a from the dye 2a
Quantum mechanic calculations of 3a also predict a hypsochromic shift of the absorption band of about 20 nm. The conformational flexibility of this compound can be studied from the potential energy curve for the compound without substituents (Fig. 6), and considering the rotation around the connecting single bond. The most stable conformation has the two pyrol units coplanar, in the so-called Z-syn form (dihedral angle 0°, Fig. 6), likely stabilized by an NH…N intramolecular H-bond. The Z-anti conformation (dihedral angle 180°, Fig. 6), where this bond cannot be formed, must be less stable. The Z-syn form has an extra stabilization of 8.3 kcal mol−1 with respect to the Z-anti conformation. The energy barrier for the change Z-syn→Z-anti transition is 22.7 kcal mol−1. This relatively high-energy barrier should be a consequence of the single/double bond character of the methene bridge group linking the two pyrrol moieties, owing to the two extreme tautomeric structures of this system (Scheme 2). A detailed conformational study of dipyrromethene systems has been published elsewhere [39]. Our results suggest the important role of the BF2 bridge to achieve a rigid structure and, hence, highly fluorescent systems.

Conformational potential energy curve of the dipyrromethene core calculated by the B3LYP/6–31G method
Taking into account the observed laser behavior of these dyes [30], it can be established a nice correlation between the fluorescence properties and the lasing characteristics of the 3-acetamido dyes 2a and 2b. Indeed, both the emission and lasing bands are hypsochromically shifted with the solvent polarity, and the emission and lasing efficiencies are nearly solvent independent. However, dye 1a does not lase, in spite of having similar fluorescence quantum yield. The photophysical properties registered at 10−3 M, a concentration of the same order as that employed to observe the lasing action, showed non-remarkable changes (i.e., absence of dye-aggregation), apart from those induced in the fluorescence properties arising from the reabsorption and reemission phenomena [40]. Once these effects were corrected, the photophysical properties were similar to those recorded in more diluted solutions. Therefore, for the moment we have not a plausible explanation for the absence of laser emission in 1a. Further work is in progress in order to find this explanation.
From the above discussion, it can be concluded that the broadening of the absorption band observed mainly for the 3-amino dye 1a in 2,2,2-trifluoroethanol should be assigned to the high acidity of this solvent. Indeed, the broad absorption bands recorded for the same dye in this solvent (Fig. 3, curve c) reminds those observed in acidic ethanol solutions (Fig. 4, curves of solutions with [H+] in the range 10−2 to 10−4 M). Both, the hypsochromic shift of the absorption band and, mainly, the drastic loss of the fluorescence of 1a in acid media allows determining the acidity of the surrounding environment by the photophysical behavior of this dye.
Conclusions
The presence of a 3-amino or 3-acetamido substituent in the BODIPY core in the dyes 1a, 2a, and 2b induces important changes in their photophysical properties in polar/protic media, with regard to the presence of alkyl groups. The acetamido group in 2a and 2b leads to an extended π-system and, hence, to a bathochromic shift of the spectral bands, with efficient fluorescence and lasing emission. On the other hand, in acid media the absorption band of the 3-amino dye 1a shifts to higher energies and becomes broader. In high acidity media, the BODIPY system of the three dyes loses its BF2 linking unit, giving rise to the corresponding non-fluorescent dipyrromethene compounds. In the particular case of 1a, the dependence of its photophysics on the solvent acidity could allow the use of this molecule as a fluorescent sensor of environmental acidity.
References
Boyer JH, Haag AM, Sathyamoorthi G, Soong M-L, Tsangaris K, Pavlopoulos TG (1993) Pyrromethene-BF2 complexes as laser dyes: 2. Heteroat Chem 4:39–49
Yariv E, Schultheiss S, Saraidarov T, Reisfeld R (2001) Efficiency and photostability of dye-doped solid-state lasers in different hosts. Opt Mater 16:29–38
Pavlopoulos TG (2002) Scaling of dye lasers with improved laser dyes. Prog Quantum Electron 26:193–224
Guilford J II, Huang Z, Pacheco D (2003) Fluorescence and lasing properties of meso-substituted benzo-fused pyrromethene dyes in solid host media. SPIE-Int Soc Opt Eng 4968:24–34
Costela A, García-Moreno I, Sastre R (2003) Polymeric solid-state dye lasers: recent developments. Phys Chem Chem Phys 5:4745–4763
Qin W, Baruah M, Van der Auweraer M, De Schryver FC, Boens N (2005) Photophysical properties of borondipyrromethene analogues in solution. J Phys Chem A 109:7371–7384
Kee HL, Kirmaier C, Yu L, Thamyongkit P, Youngbllod WJ, Calder ME, Ramos L, Noll BC, Bocian DF, Scheidt WR, Birge RR, Lindsey JS, Holten D (2005) Structural control of the photodynamics of boron-dipyrrin complexes. J Phys Chem B 109:20433–20443
López Arbeloa F, Bañuelos J, Martínez V, Arbeloa T, López Arbeloa I (2005) Structural, photophysical and lasing properties of pyrromethene dyes. Int Rev Phys Chem 24:339–374
Jones G II, Klueva O, Kumar S, Pacheco D (2001) Photochemical and lasing properties of pyrromethene dyes. SPIE-Int Soc Opt Eng 4267:24–35
Sisk WN, Ono N, Yano T, Wada M (2002) Photostability studies of three new bicycle-boron dipyrromethene difuoride dyes. Dyes Pigm 55:143–150
Yang Y, Wang M, Qian G, Wang Z, Fan X (2004) Laser properties and photostabilities of laser dyes doped in ORMOSILs. Opt Mater 24:621–628
Alvarez M, Amat-Guerri F, Campo L, Costela A, García-Moreno I, García O, Gómez C, Liras M, Sastre R (2004) Solid-state dye lasers based on dipyrromethene dyes: recent developments. Recent Res Devel Appl Phys 7:19–40
Metzker ML, Lu J, Gibbs RA (1996) Electrophoretically uniform fluorescent dyes for automated DNA sequencing. Science 271:1420–1422
Farber SA, Pack M, Ho S-Y, Johnson ID, Wagner DS, Dosch R, Mullins MC, Hendrickson HS, Hendrickson EK, Halpern ME (2001) Genetic analysis of digestive physiology using fluorescent phospholipids reporters. Science 292:1385–1388
Reents R, Wagner M, Kuhlmann J, Waldmann H (2004) Synthesis and application of fluorescence-labeled ras-proteins for live-cell imaging. Angew Chem Int Ed 43:2711–2714
Lammi RK, Wagner RW, Ambroise A, Diers JR, Bocian DF, Holten D, Lindsey JS (2001) Rapid energy transfer in cascade-type bodipy dyes. J Phys Chem B 105:5341–5352
Wan C-W, Burghart A, Chen J, Bergström F, Johansson LBA, Wolford MF, Kim TG, Topp MR, Hochtrasser RM, Burgess K (2003) Anthracene-BODIPY cassettes: synthesis and energy transfer. Chem Eur J 9:4430–4441
D’Souza F, Smith PM, Zandler ME, McCarthy AL, Itou M, Araki Y, Ito O (2004) Energy transfer followed by electron transfer in a supramolecular triad composed of boron dipyrrin, zinc porphyrin, and fullerene: a model for the photosynthetic antenna-reaction center complex. J Am Chem Soc 126:7898–7907
Harriman A, Izzet G, Ziessel R (2006) Rapid energy transfer in cascade-type bodipy dyes. J Am Chem Soc 128:10231–10239
Costela A, García-Moreno I, Gomez C, Sastre R, Amat-Guerri F, Liras M, López Arbeloa F, Bañuelos J, López Arbeloa I (2002) Photophysical and lasing properties of new analogs of the boron-dipyrromethene laser dye PM567 in liquid solution. J Phys Chem A 106:7736–7742
Lai RY, Bard AJ (2003) Electrogenerated chemiluminescence 71. Photophysical, electrochemical, and electrogenerated chemiluminescent properties of selected dipyrromethene-BF2 dyes. J Phys Chem B 107:5036–5042
García-Moreno I, Costela A, Campo L, Sastre R, Amat-Guerri F, Liras M, López Arbeloa F, Bañuelos J, López Arbeloa I (2004) 8-phenyl-substituted dipyrromethene∙BF2 complexes as highly efficient and photostable laser dyes. J Phys Chem A 108:3315–3323
Shen Z, Röhr H, Rurack K, Uno H, Spieles M, Schulz B, Reck G, Ono N (2004) Boron-diindomethene (BDI) dyes and their tetrahydrobicyclo precursors: a route to a new class of highly emissive fluorophores for the red spectral range. Chem Eur J 10:4853–4871
Rohand T, Qin W, Boens N, Dehaen W (2006) Palladium-catalyzed coupling reaction s for the functionalization of BODIPY dyes fluorescence spanning the visible spectrum. Eur J Org Chem 2006(20):4658–4663
Bañuelos J, López Arbeloa F, Martínez V, López Arbeloa I (2004) Theoretical study of the ground and excited electronic states of pyrromethene 546 laser dye and related compounds. Chem Phys 296:13–22
Quartorolo AD, Russo N, Sicilia E (2006) Structures and electronic absorption spectra of a recently synthesized class of photodynamic therapy agents. Chem Eur J 12:6797–6803
de Silva AP, Gunaratne N, Gunnlaugsson T, Huxley AJM, McCoy CP, Rademacher JT, Rice TE (1997) Signaling recognition events with fluorescent sensors and switches. Chem Rev 97:1515–1556
Valeur B, Leray I (2000) Design principles of fluorescent molecular sensors for cation recognition. Coord Chem Rev 205:3–40
Rurack K, Resch-Genger U (2002) Rigidization, preorientation and electronic decoupling—‘the magic triangle’ for the design of highly efficient fluorescent sensors and switches. Chem Soc Rev 31:116–127
Liras M, Bañuelos Prieto J, Pintado-Sierra M, López Arbeloa F, García-Moreno I, Costela A, Infantes L, Sastre R, Amat-Guerri F (2007) Synthesis, photophysical properties, and laser behavior of 3-amino and 3-acetamido BODIPY dyes. Org Lett 9:4183–4186
López Arbeloa F, López Arbeloa T, López Arbeloa I, García-Moreno I, Costela A, Sastre R, Amat-Guerri F (1998) Photophysical and lasing properties of pyrromethene 567 dye in liquid solution. Environment effects. Chem Phys 236:331–341
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian-03. Gaussian Inc, Pittsburgh PA
Bañuelos J, López Arbeloa F, Martínez V, Arbeloa López T, López Arbeloa I (2004) Photophysical properties of the pyrromethene 597 dye: solvent effect. J Phys Chem A 108:5503–5508
Badré S, Monnier V, Méallet-Renault R, Dumas-Verdes C, Schmidt EY, Mikhaleva AI, Laurent G, Levi G, Ibanez A, Trofimov BA, Pansu RB (2006) Fluorescence of molecular micro- and nanocristals prepared with bodipy derivatives. J Photochem Photobiol A 183:238–246
Kollmannsberger M, Rurack K, Resch-Genger U, Daub J (1998) Ultrafast charge transfer in amino-substituted boron dipyrromethene dyes and its inhibition by cation complexation: a new design for highly sensitive fluorescent probes. J Phys Chem A 102:10211–10220
López Arbeloa F, López Arbeloa T, López Arbeloa I (1994) Photophysics of amino-aromatic laser dyes. Trends Photochem Photobiol 3:145–155
López Arbeloa F, López Arbeloa T, López Arbeloa I (2001) In Nalwa HS (Ed.) Handbook of Advanced Electronic and Photonic Materials and Devices, Academic Press, San Diego, CA, vol. 7, pp. 209–245
Reisfeld R, Yariv E, Minti H (1997) New developments in solid state lasers. Opt Mater 8:31–36
Korkin A, Mark F, Schaffner K, Gorb L, Leszczynski J (1996) Theoretical ab initio and semiempirical studies of biologically important di- and oligopyrrolic compounds; pyrromethene and protonated pyrromethene. J Molec Struct 388:121–137
López Arbeloa I (1980) Fluorescence quantum yield evaluation: corrections for re-absorption and re-emission. J Photochem 14:97–105
Acknowledgements
This work has been supported by Spanish Ministerio de Educación y Ciencia (MEC; Project MAT2004-04643-C03-01 and -02). Computational resources were provided by the “ARINA” SGI/IZO-SGIker Service at the Basque Country University, supported by the Spanish MEC and the European Social Fund. ML acknowledges a “Juan de la Cierva” contract from MEC. Sandra Salleres thanks to UPV/EHU for a predoctoral fellowship.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bañuelos, J., López Arbeloa, F., Arbeloa, T. et al. Photophysical Characterization of New 3-Amino and 3-Acetamido BODIPY Dyes with Solvent Sensitive Properties. J Fluoresc 18, 899–907 (2008). https://doi.org/10.1007/s10895-008-0320-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10895-008-0320-7